Department of Dermatology, Affiliated Hospital of Qingdao University, Qingdao, Shandong, China (mainland).
Department of Dermatology, The First Affiliated Hospital of Nanchang University, Nanchang, Jiangxi, China (mainland).
Med Sci Monit. 2019 Apr 20;25:2896-2907. doi: 10.12659/MSM.913881.
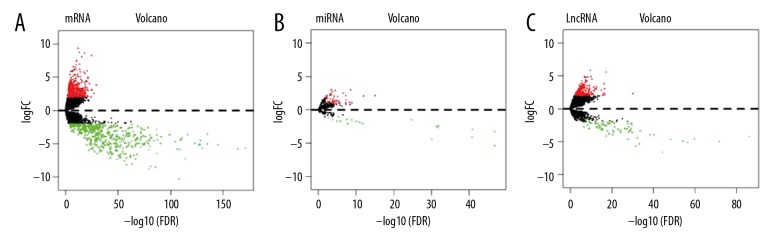
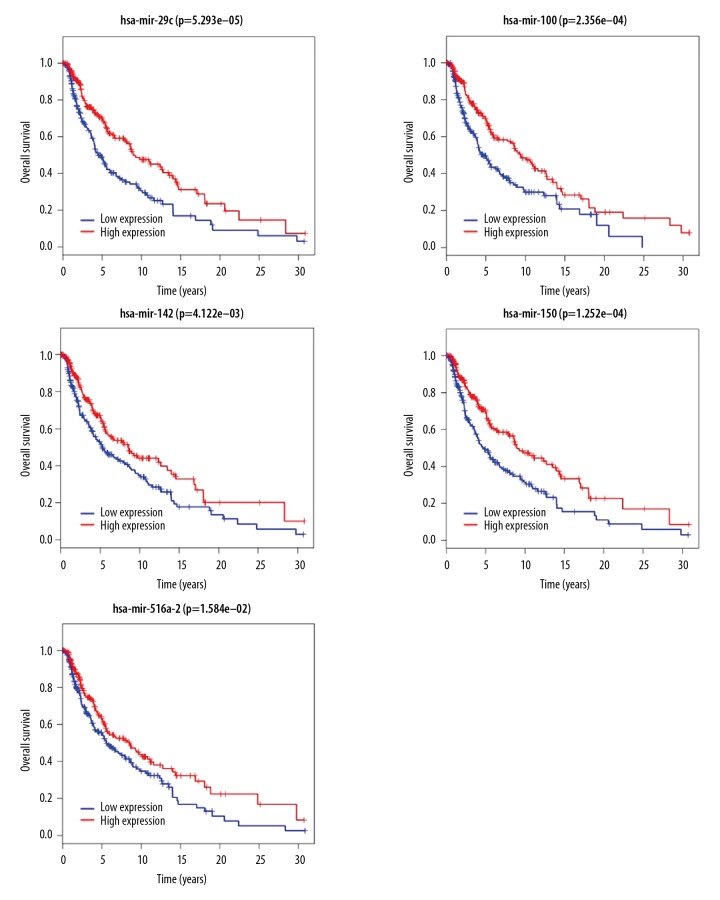
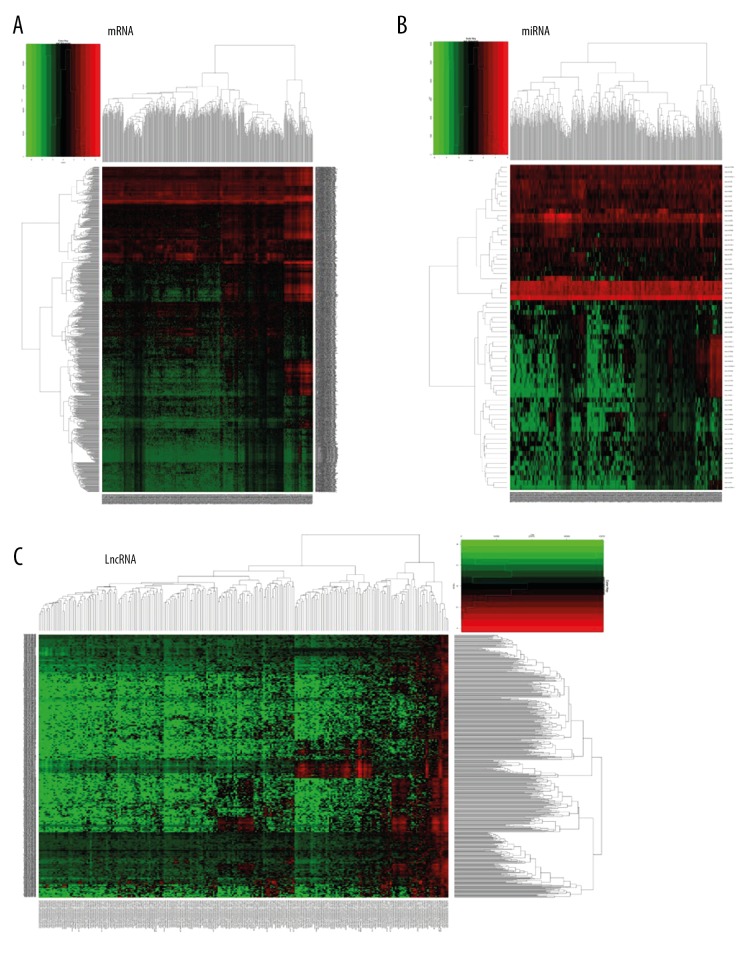
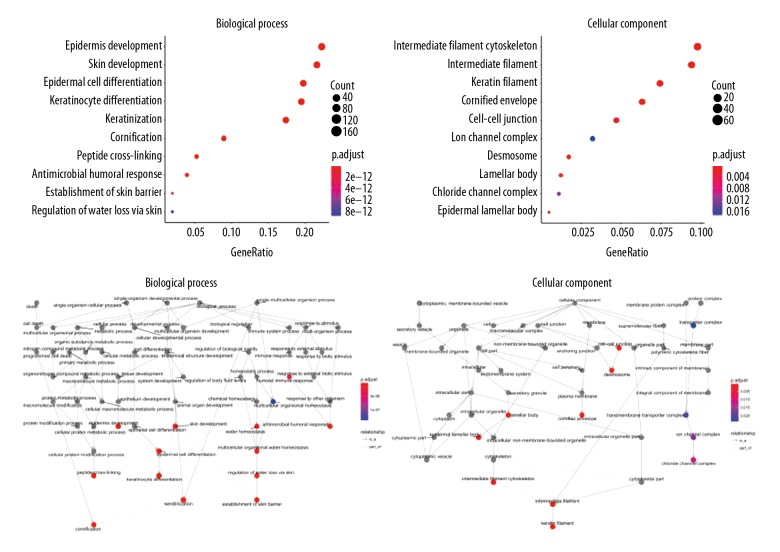
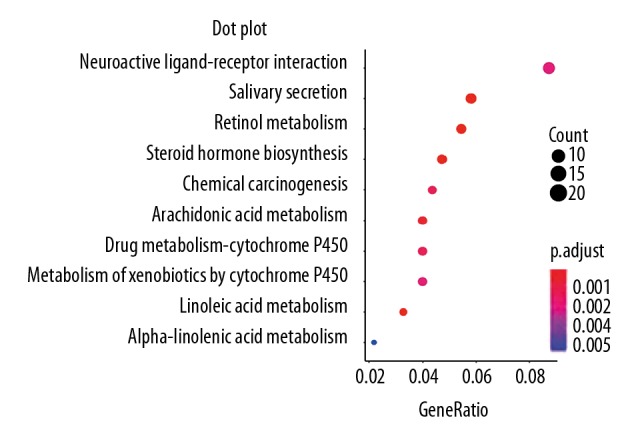
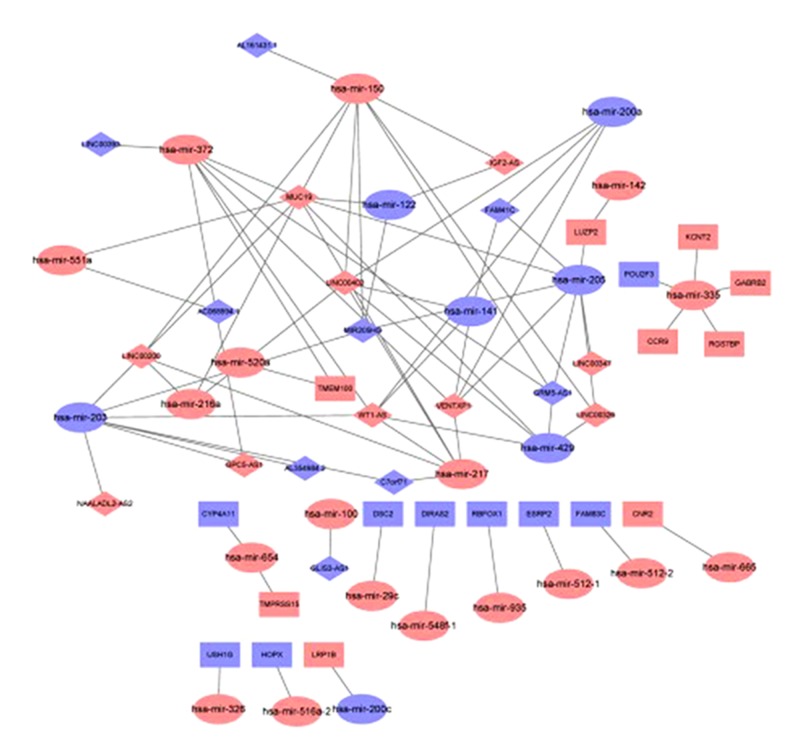
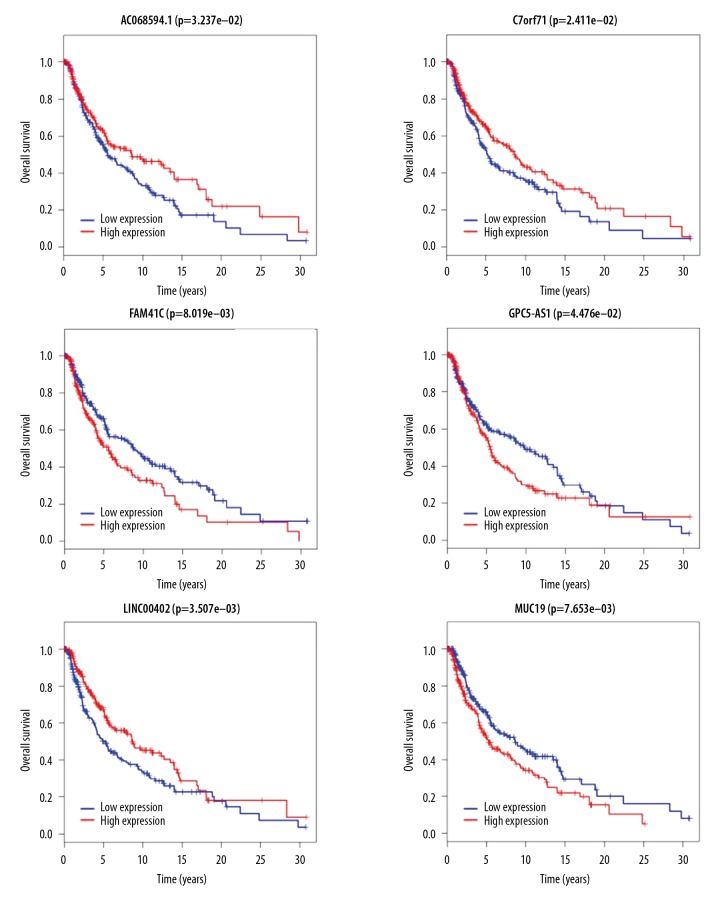
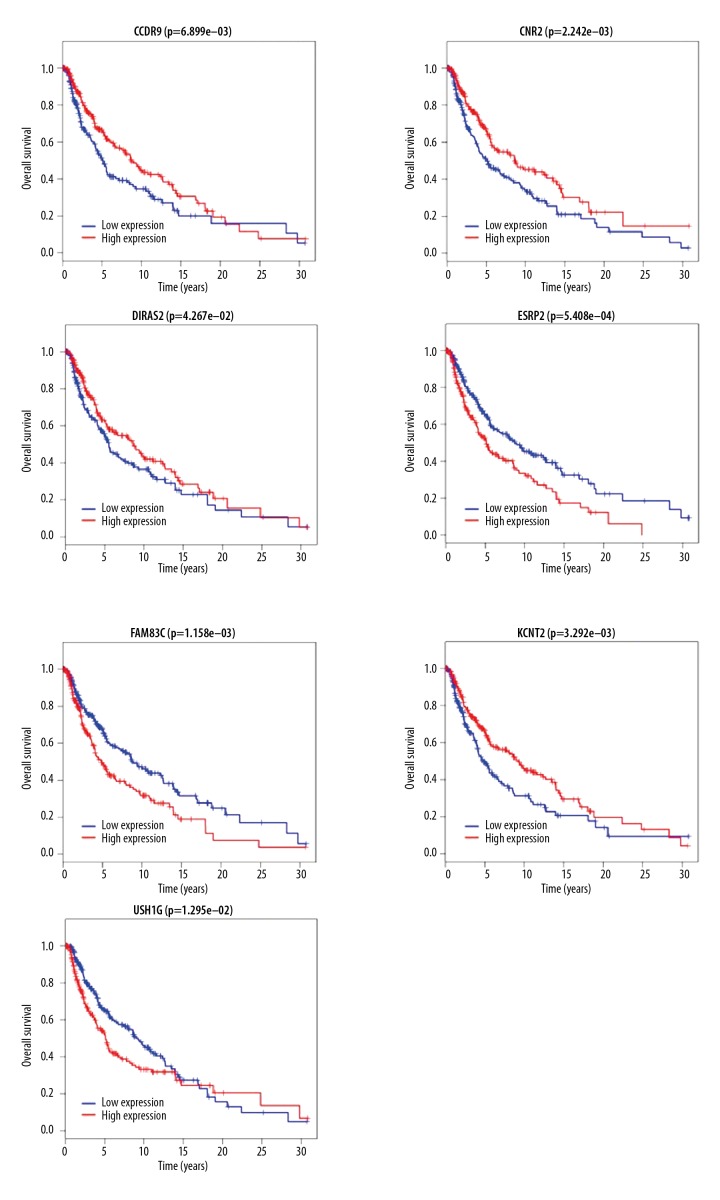
Worldwide, metastatic melanoma of the skin has an aggressive course with high morbidity and mortality. Therefore, an increased understanding of the pathogenesis of metastatic melanoma has gained increasing attention, including the role of epigenetic modification and competing endogenous RNA (ceRNA). This study aimed to used bioinformatics data to undertake an integrative analysis of long noncoding RNA (lncRNA), microRNA (miRNA) and mRNA expression to construct a ceRNA network in metastatic melanoma. Data from the Cancer Genome Atlas (TCGA), the Gene Ontology (GO) database, and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway were analyzed. There were 471 cases that included 103 primary solid tumors and 368 cases of metastatic melanoma that included transcriptome sequencing data (including lncRNA and mRNA); 452 cases had miRNA sequencing data. Analysis of chip data identified 85 6 mRNAs, 67 miRNAs, and 250 lncRNAs that were differentially expressed in cases of metastatic melanoma, of which 25 miRNAs, 18 lncRNAs, and 18 mRNAs participated in the formation of ceRNAs. Survival analysis identified seven differentially expressed mRNAs, five differentially expressed miRNAs (miRNA-29c, miRNA-100, miR-142-3p, miR-150, miR-516a-2), and six differentially expressed lncRNAs (AC068594.1, C7orf71, FAM41C, GPC5-AS1, MUC19, LINC00402) that were correlated with survival time in patients with metastatic melanoma. Bioinformatics data and integrative analysis identified lncRNA, miRNA, and mRNA expression to construct a ceRNA and patient survival network in metastatic melanoma. These findings support the need for further studies on the mechanisms involved in the regulation of metastatic melanoma by ceRNAs.
全球范围内,皮肤转移性黑色素瘤具有侵袭性病程,发病率和死亡率均较高。因此,人们越来越关注对转移性黑色素瘤发病机制的深入了解,包括表观遗传修饰和竞争内源性 RNA(ceRNA)的作用。本研究旨在利用生物信息学数据对长链非编码 RNA(lncRNA)、微小 RNA(miRNA)和信使 RNA(mRNA)表达进行综合分析,构建转移性黑色素瘤的 ceRNA 网络。分析了癌症基因组图谱(TCGA)、基因本体论(GO)数据库和京都基因与基因组百科全书(KEGG)通路的数据。有 471 例病例,包括 103 例原发性实体瘤和 368 例转移性黑色素瘤,包括转录组测序数据(包括 lncRNA 和 mRNA);452 例有 miRNA 测序数据。芯片数据分析鉴定出 856 个 mRNAs、67 个 miRNAs 和 250 个 lncRNAs 在转移性黑色素瘤病例中表达差异,其中 25 个 miRNAs、18 个 lncRNAs 和 18 个 mRNAs参与了 ceRNA 的形成。生存分析鉴定出 7 个差异表达的 mRNAs、5 个差异表达的 miRNAs(miRNA-29c、miRNA-100、miR-142-3p、miR-150、miR-516a-2)和 6 个差异表达的 lncRNAs(AC068594.1、C7orf71、FAM41C、GPC5-AS1、MUC19、LINC00402)与转移性黑色素瘤患者的生存时间相关。生物信息学数据和综合分析鉴定出 lncRNA、miRNA 和 mRNA 表达,构建了转移性黑色素瘤的 ceRNA 和患者生存网络。这些发现支持进一步研究 ceRNA 调节转移性黑色素瘤的机制的必要性。