Katuwawala Akila, Peng Zhenling, Yang Jianyi, Kurgan Lukasz
Department of Computer Science, Virginia Commonwealth University, USA.
Center for Applied Mathematics, Tianjin University, Tianjin, China.
Comput Struct Biotechnol J. 2019 Mar 26;17:454-462. doi: 10.1016/j.csbj.2019.03.013. eCollection 2019.
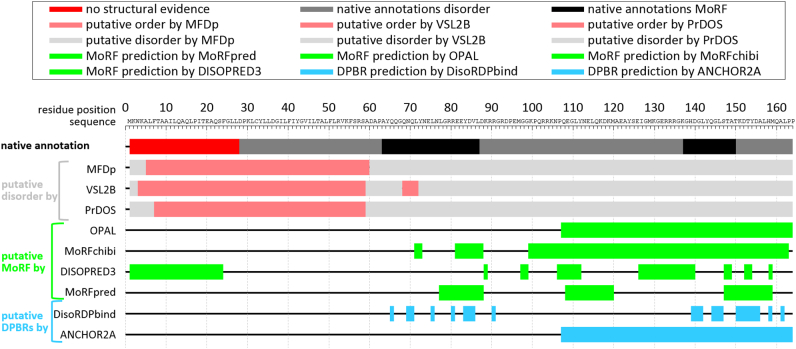
Molecular recognition features (MoRFs) are short protein-binding regions that undergo disorder-to-order transitions (induced folding) upon binding protein partners. These regions are abundant in nature and can be predicted from protein sequences based on their distinctive sequence signatures. This first-of-its-kind survey covers 14 MoRF predictors and six related methods for the prediction of short protein-binding linear motifs, disordered protein-binding regions and semi-disordered regions. We show that the development of MoRF predictors has accelerated in the recent years. These predictors depend on machine learning-derived models that were generated using training datasets where MoRFs are annotated using putative disorder. Our analysis reveals that they generate accurate predictions. We identified eight methods that offer area under the ROC curve (AUC) ≥ 0.7 on experimentally-validated test datasets. We show that modern MoRF predictors accurately find experimentally annotated MoRFs even though they were trained using the putative disorder annotations. They are relatively highly-cited, particularly the methods available as webservers that on average secure three times more citations than methods without this option. MoRF predictions contribute to the experimental discovery of protein-protein interactions, annotation of protein functions and computational analysis of a variety of proteomes, protein families, and pathways. We outline future development and application directions for these tools, stressing the importance to develop novel tools that would target interactions of disordered regions with other types of partners.
分子识别特征(MoRFs)是短的蛋白质结合区域,在与蛋白质伴侣结合时会经历无序到有序的转变(诱导折叠)。这些区域在自然界中很丰富,可以根据其独特的序列特征从蛋白质序列中预测出来。这项首创的调查涵盖了14种MoRF预测器以及6种用于预测短蛋白质结合线性基序、无序蛋白质结合区域和半无序区域的相关方法。我们表明,近年来MoRF预测器的发展加速了。这些预测器依赖于机器学习衍生的模型,这些模型是使用训练数据集生成的,在这些数据集中,MoRFs是使用假定的无序进行注释的。我们的分析表明它们能产生准确的预测。我们确定了8种方法,在经过实验验证的测试数据集上,其ROC曲线下面积(AUC)≥0.7。我们表明,现代MoRF预测器即使是使用假定的无序注释进行训练,也能准确找到经过实验注释的MoRFs。它们的引用率相对较高,特别是那些作为网络服务器可用的方法,平均而言,其引用次数是没有此选项方法的三倍多。MoRF预测有助于蛋白质 - 蛋白质相互作用的实验发现、蛋白质功能的注释以及对各种蛋白质组、蛋白质家族和途径的计算分析。我们概述了这些工具未来的发展和应用方向,强调开发针对无序区域与其他类型伴侣相互作用的新型工具的重要性。