Okur Volkan, LeDuc Charles A, Guzman Edwin, Valivullah Zaheer M, Anyane-Yeboa Kwame, Chung Wendy K
Department of Pediatrics, Columbia University, New York, New York 10032, USA.
Department of Pediatrics, Naomi Berrie Diabetes Center, Columbia University, New York, New York 10032, USA.
Cold Spring Harb Mol Case Stud. 2019 Jun 3;5(3). doi: 10.1101/mcs.a004101. Print 2019 Jun.
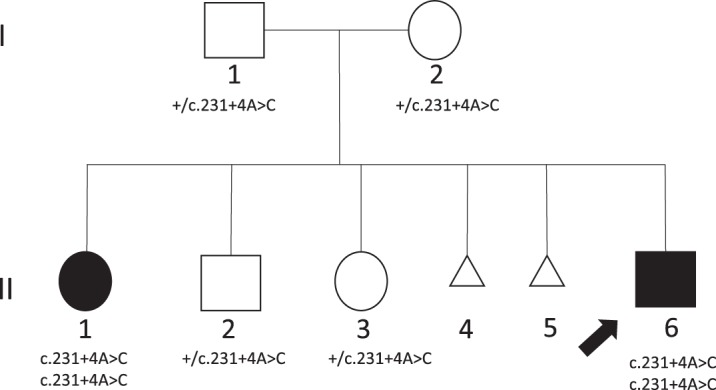
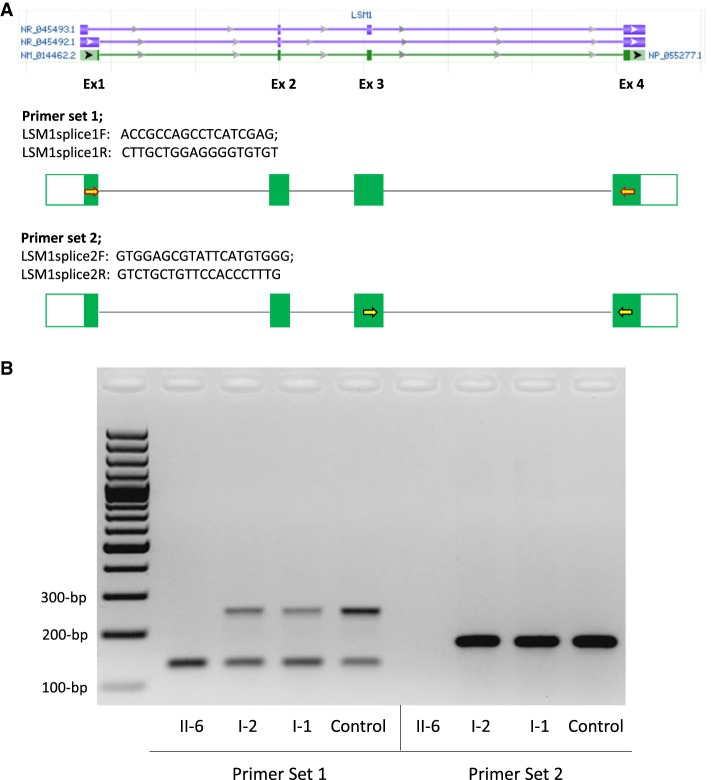
Two siblings, one male and one female, ages 6 and 13 yr old, have similar clinical features of global developmental delay, multiple congenital anomalies affecting the cardiac, genitourinary, and skeletal systems, and abnormal eye movements. Whole-genome sequencing revealed a homozygous splice variant (NM_014462.3:c.231+4A>C) in that segregated with the phenotype in the family. LSM1 has a role in pre-mRNA splicing and degradation. Expression studies revealed absence of expression of the canonical isoform in the affected individuals. The knockout mice have a partially overlapping phenotype that affects the brain, heart, and eye. To our knowledge, has not been associated with any human disorder; however, the tissue expression pattern, gene constraint, and the similarity of the phenotype in our patients and the knockout mice models suggest it has a role in the development of multiple organ systems in humans.
两名兄弟姐妹,一男一女,年龄分别为6岁和13岁,具有相似的临床特征,包括全面发育迟缓、影响心脏、泌尿生殖系统和骨骼系统的多种先天性异常以及异常的眼球运动。全基因组测序显示在该基因中存在一个纯合剪接变体(NM_014462.3:c.231+4A>C),该变体在家族中与表型共分离。LSM1在mRNA前体剪接和降解中起作用。表达研究显示在受影响个体中缺乏典型异构体的表达。该基因敲除小鼠具有部分重叠的表型,影响大脑、心脏和眼睛。据我们所知,该基因尚未与任何人类疾病相关联;然而,组织表达模式、基因限制以及我们患者与基因敲除小鼠模型中表型的相似性表明它在人类多器官系统发育中起作用。