Department of Pediatrics and Clinical Genetics, GROW-School for Oncology and Developmental Biology, Maastricht University Medical Centre, P. Debyelaan 25, P.O. Box 5800, 6202 AZ, Maastricht, The Netherlands.
Amsterdam UMC, University of Amsterdam, Pediatric Metabolic Diseases, Emma Children's Hospital, Amsterdam, Netherlands.
Orphanet J Rare Dis. 2019 Apr 27;14(1):86. doi: 10.1186/s13023-019-1047-z.
Classic galactosemia is a rare inborn error of carbohydrate metabolism, caused by a severe deficiency of the enzyme galactose-1-phosphate uridylyltransferase (GALT). A galactose-restricted diet has proven to be very effective to treat the neonatal life-threatening manifestations and has been the cornerstone of treatment for this severe disease. However, burdensome complications occur despite a lifelong diet. For rare diseases, a patient disease specific registry is fundamental to monitor the lifespan pathology and to evaluate the safety and efficacy of potential therapies. In 2014, the international Galactosemias Network (GalNet) developed a web-based patient registry for this disease, the GalNet Registry. The aim was to delineate the natural history of classic galactosemia based on a large dataset of patients.
Observational data derived from 15 countries and 32 centers including 509 patients were acquired between December 2014 and July 2018.
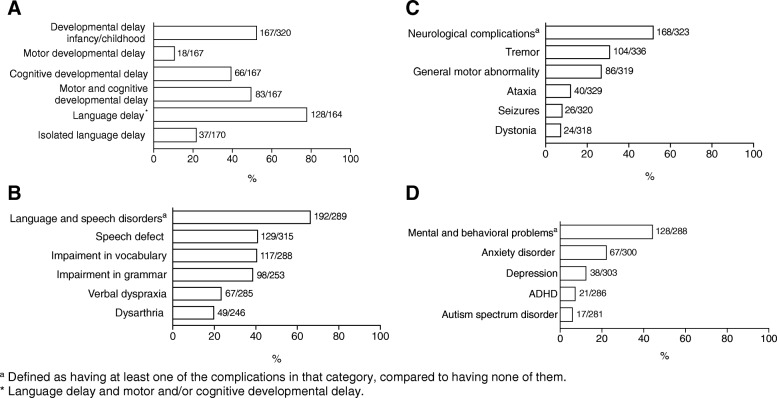
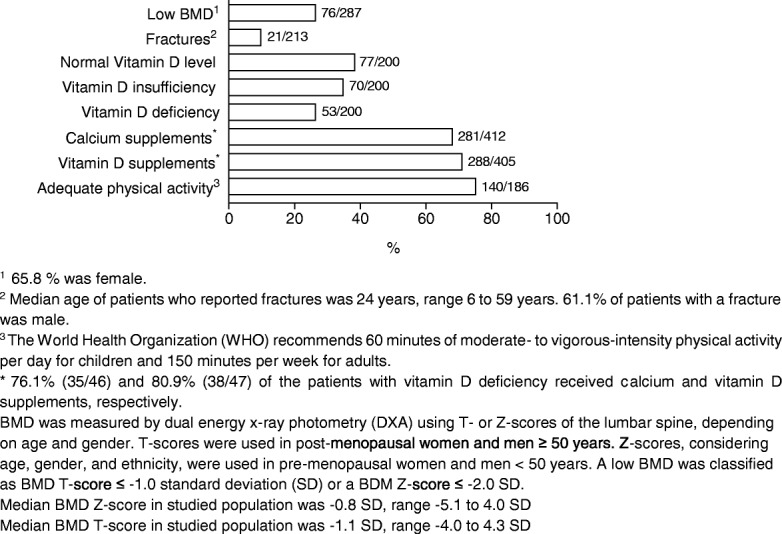
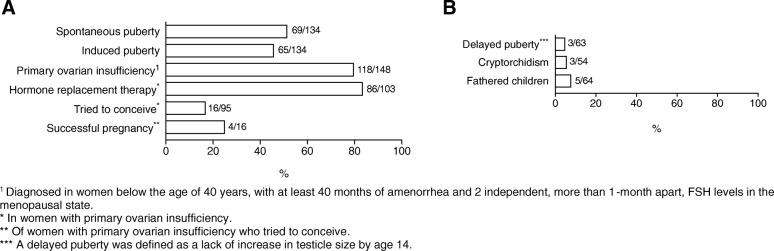
Most affected patients experienced neonatal manifestations (79.8%) and despite following a diet developed brain impairments (85.0%), primary ovarian insufficiency (79.7%) and a diminished bone mineral density (26.5%). Newborn screening, age at onset of dietary treatment, strictness of the galactose-restricted diet, p.Gln188Arg mutation and GALT enzyme activity influenced the clinical picture. Detection by newborn screening and commencement of diet in the first week of life were associated with a more favorable outcome. A homozygous p.Gln188Arg mutation, GALT enzyme activity of ≤ 1% and strict galactose restriction were associated with a less favorable outcome.
This study describes the natural history of classic galactosemia based on the hitherto largest data set.
经典型半乳糖血症是一种罕见的碳水化合物代谢先天性缺陷,由半乳糖-1-磷酸尿苷酰转移酶(GALT)严重缺乏引起。限制半乳糖饮食已被证明对治疗危及生命的新生儿期表现非常有效,是治疗这种严重疾病的基石。然而,尽管患者进行了终生饮食限制,仍会出现严重的并发症。对于罕见疾病,患者特定疾病登记对于监测寿命病理学以及评估潜在治疗方法的安全性和疗效至关重要。2014 年,国际半乳糖血症网络(GalNet)为该疾病开发了一个基于网络的患者登记处,即 GalNet 登记处。其目的是基于大量患者数据来描绘经典半乳糖血症的自然病史。
2014 年 12 月至 2018 年 7 月,从 15 个国家和 32 个中心收集了 15 国和 32 个中心的观察性数据,包括 509 名患者。
大多数受影响的患者表现为新生儿期表现(79.8%),尽管遵循饮食治疗,仍会出现脑损伤(85.0%)、原发性卵巢功能不全(79.7%)和骨矿物质密度降低(26.5%)。新生儿筛查、开始饮食治疗的年龄、半乳糖限制饮食的严格程度、p.Gln188Arg 突变和 GALT 酶活性影响临床表型。通过新生儿筛查和在生命的第一周开始饮食与更好的结局相关。纯合 p.Gln188Arg 突变、GALT 酶活性≤1%和严格的半乳糖限制与更差的结局相关。
本研究基于迄今为止最大的数据集描述了经典型半乳糖血症的自然病史。