Heinzen R A, Mallavia L P
Infect Immun. 1987 Apr;55(4):848-55. doi: 10.1128/iai.55.4.848-855.1987.
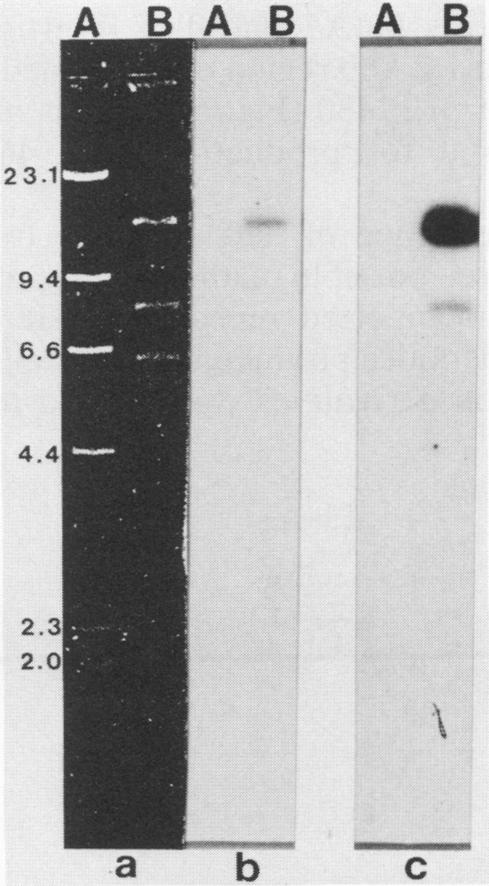
The citrate synthase gene from the obligate intracellular rickettsial parasite Coxiella burnetii was cloned and expressed in Escherichia coli. Transduction into E. coli with a C. burnetii gene library constructed in the cosmid vector pHK17 resulted in the functional complementation of the gltA mutation of E. coli MOB154. A GltA+ clone carrying 16.4 kilobase pairs of C. burnetii DNA and designated pJCC959 was isolated and characterized. Southern hybridization analysis confirmed that the pJCC959 cloned insert consists of C. burnetii DNA and that homology exists with the Rickettsia prowazekii citrate synthase gene. Subcloning analysis with the multicopy expression vector pUC8 revealed that citrate synthase expression was under control of a C. burnetti promoter. In vitro transcription-translation of subclones pLPM20 and pLPM30 established a molecular weight of ca. 46,000 for the monomer form of the cloned enzyme. Transposon Tn5 mutagenesis of pLPM30 defined the coding region to approximately 1.2 kilobase pairs of C. burnetii DNA. Maxicell analysis of selected pLPM30::Tn5 insertion derivatives identified the direction of transcription and the relative translational start and stop sites and substantiated the molecular weight value calculated from the in vitro analysis. Inhibition studies showed that citrate synthase activity in crude cell extracts obtained from strain MOB154 transformed with the cloned C. burnetii gene was markedly inhibited by 4 mM ATP, while 4 mM alpha-ketoglutarate had virtually no effect. These data indicate that the C. burnetii enzyme displays regulatory behavior characteristic of the small gram-positive bacterial and eucaryotic enzyme.
专性细胞内立克次氏体寄生虫伯氏考克斯体的柠檬酸合酶基因被克隆并在大肠杆菌中表达。用构建于黏粒载体pHK17中的伯氏考克斯体基因文库转导大肠杆菌,导致大肠杆菌MOB154的gltA突变得到功能互补。分离并鉴定了一个携带16.4千碱基对伯氏考克斯体DNA的GltA+克隆,命名为pJCC959。Southern杂交分析证实pJCC959克隆插入片段由伯氏考克斯体DNA组成,且与普氏立克次体柠檬酸合酶基因存在同源性。用多拷贝表达载体pUC8进行亚克隆分析表明,柠檬酸合酶的表达受伯氏考克斯体启动子的控制。亚克隆pLPM20和pLPM30的体外转录-翻译确定了克隆酶单体形式的分子量约为46,000。pLPM30的转座子Tn5诱变将编码区域定位到约1.2千碱基对的伯氏考克斯体DNA。对选定的pLPM30::Tn5插入衍生物进行的大细胞分析确定了转录方向以及相对的翻译起始和终止位点,并证实了从体外分析计算得到的分子量值。抑制研究表明,用克隆的伯氏考克斯体基因转化的MOB154菌株粗细胞提取物中的柠檬酸合酶活性受到4 mM ATP的显著抑制,而4 mMα-酮戊二酸几乎没有影响。这些数据表明,伯氏考克斯体酶表现出小革兰氏阳性细菌和真核酶特有的调节行为。