Minchiotti Lorenzo, Caridi Gianluca, Campagnoli Monica, Lugani Francesca, Galliano Monica, Kragh-Hansen Ulrich
Department of Molecular Medicine, University of Pavia, Pavia, Italy.
Laboratory of Molecular Nephrology, Istituto Giannina Gaslini (IRCCS), Genoa, Italy.
Front Genet. 2019 Apr 17;10:336. doi: 10.3389/fgene.2019.00336. eCollection 2019.
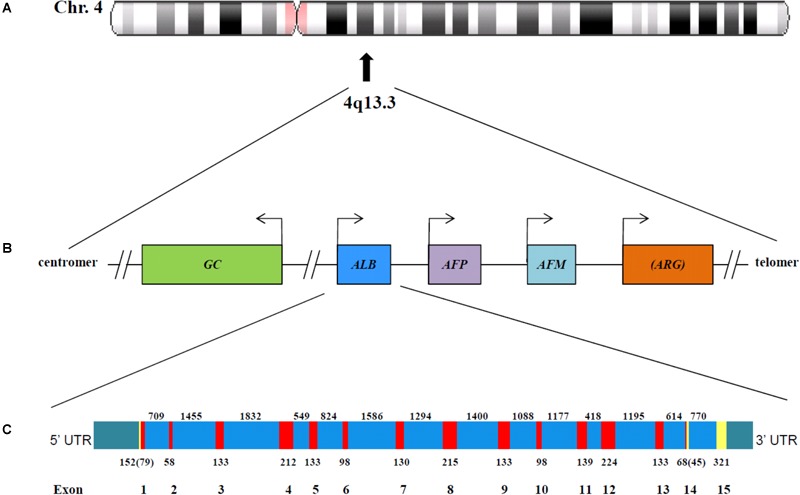
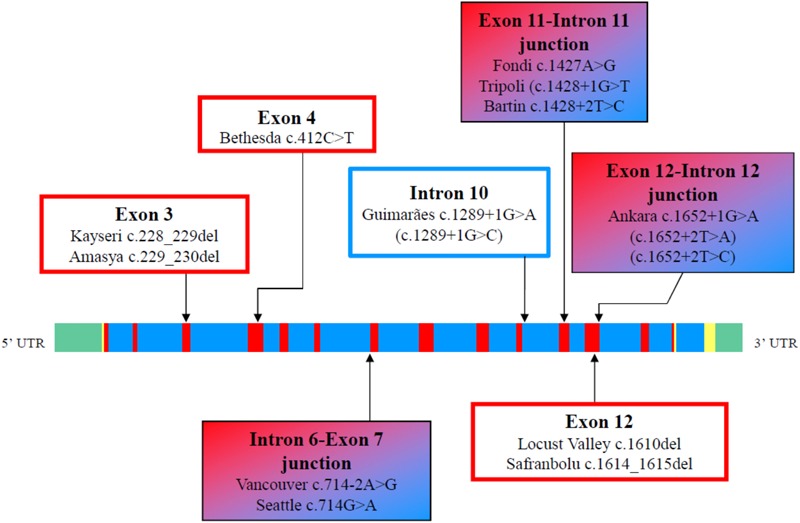
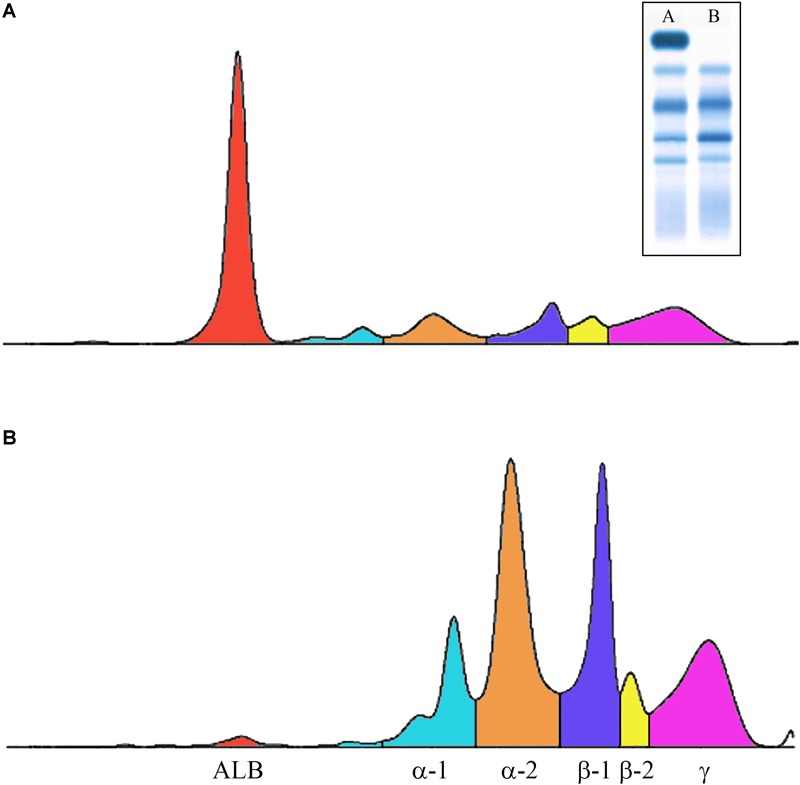
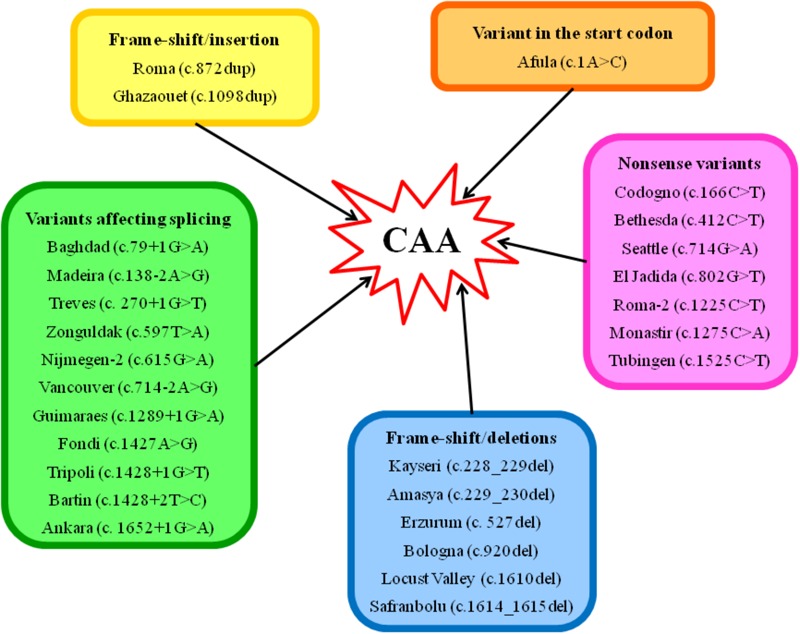
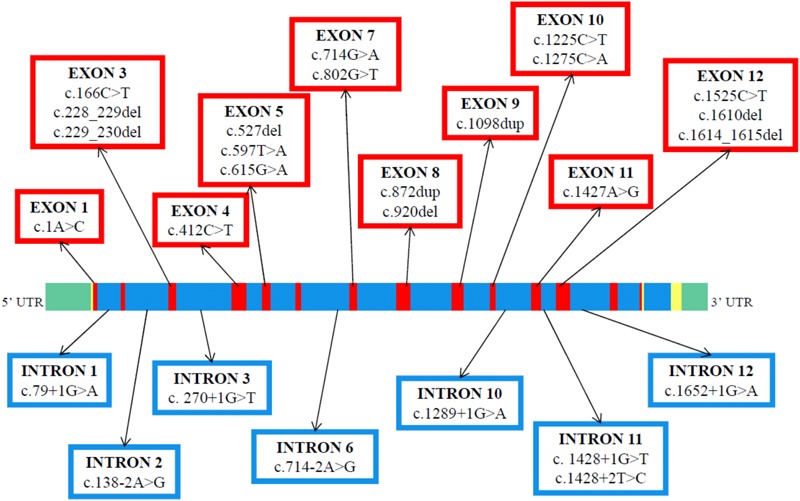
Congenital analbuminemia (CAA) is an inherited, autosomal recessive disorder with an incidence of 1:1,000,000 live birth. Affected individuals have a strongly decreased concentration, or complete absence, of serum albumin. The trait is usually detected by serum protein electrophoresis and immunochemistry techniques. However, due to the existence of other conditions in which the albumin concentrations are very low or null, analysis of the albumin () gene is necessary for the molecular diagnosis. CAA can lead to serious consequences in the prenatal period, because it can cause miscarriages and preterm birth, which often is due to oligohydramnios and placental abnormalities. Neonatally and in early childhood the trait is a risk factor that can lead to death, mainly from fluid retention and infections in the lower respiratory tract. By contrast, CAA is better tolerated in adulthood. Clinically, in addition to the low level of albumin, the patients almost always have hyperlipidemia, but they usually also have mild oedema, reduced blood pressure and fatigue. The fairly mild symptoms in adulthood are due to compensatory increment of other plasma proteins. The condition is rare; clinically, only about 90 cases have been detected worldwide. Among these, 53 have been studied by sequence analysis of the gene, allowing the identification of 27 different loss of function (LoF) pathogenic variants. These include a variant in the start codon, frame-shift/insertions, frame-shift/deletions, nonsense variants, and variants affecting splicing. Most are unique, peculiar for each affected family, but one, a frame-shift deletion called Kayseri, has been found to cause about one third of the known cases allowing to presume a founder effect. This review provides an overview of the literature about CAA, about supportive and additional physiological and pharmacological information obtained from albumin-deficient mouse and rat models and a complete and up-to-date dataset of the pathogenic variants identified in the gene.
先天性无白蛋白血症(CAA)是一种遗传性常染色体隐性疾病,活产发病率为1:1,000,000。受影响个体的血清白蛋白浓度大幅降低或完全缺失。该特征通常通过血清蛋白电泳和免疫化学技术检测。然而,由于存在其他白蛋白浓度极低或为零的情况,因此对白蛋白()基因进行分析对于分子诊断是必要的。CAA在孕期可导致严重后果,因为它可引起流产和早产,这通常是由于羊水过少和胎盘异常所致。在新生儿期和儿童早期,该特征是一个危险因素,可导致死亡,主要原因是液体潴留和下呼吸道感染。相比之下,CAA在成年期耐受性较好。临床上,除了白蛋白水平低外,患者几乎总是患有高脂血症,但通常也有轻度水肿、血压降低和疲劳。成年期症状相对较轻是由于其他血浆蛋白的代偿性增加。这种疾病很罕见;临床上,全球仅检测到约90例。其中,53例通过对基因的序列分析进行了研究,从而鉴定出27种不同的功能丧失(LoF)致病变异。这些变异包括起始密码子变异、移码/插入、移码/缺失、无义变异以及影响剪接的变异。大多数变异是独特的,每个受影响家族都有其特殊性,但有一种名为开塞利的移码缺失变异,已发现它导致了约三分之一的已知病例,这表明可能存在奠基者效应。本综述概述了有关CAA的文献、从白蛋白缺乏的小鼠和大鼠模型获得的支持性及其他生理和药理学信息,以及在该基因中鉴定出的致病变异的完整最新数据集。