Department of Ocular Oncology, Wills Eye Hospital, Philadelphia, PA, USA; Department of Ocular Oncology, Centre for Sight, Hyderabad, Telangana, India.
Department of Ocular Oncology, Wills Eye Hospital, Philadelphia, PA, USA, India.
Indian J Ophthalmol. 2019 Jun;67(6):755-762. doi: 10.4103/ijo.IJO_845_19.
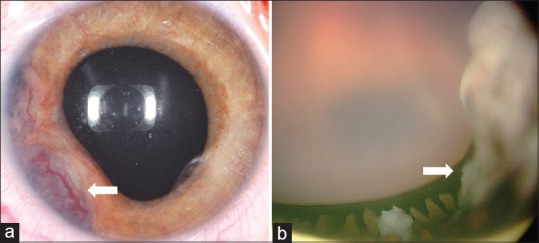
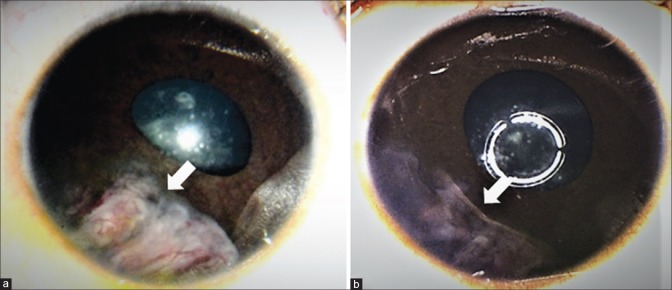
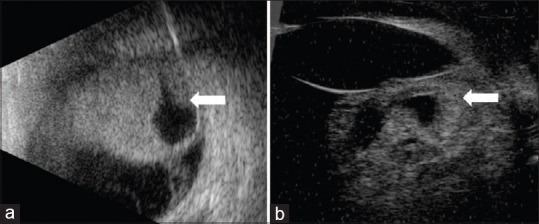
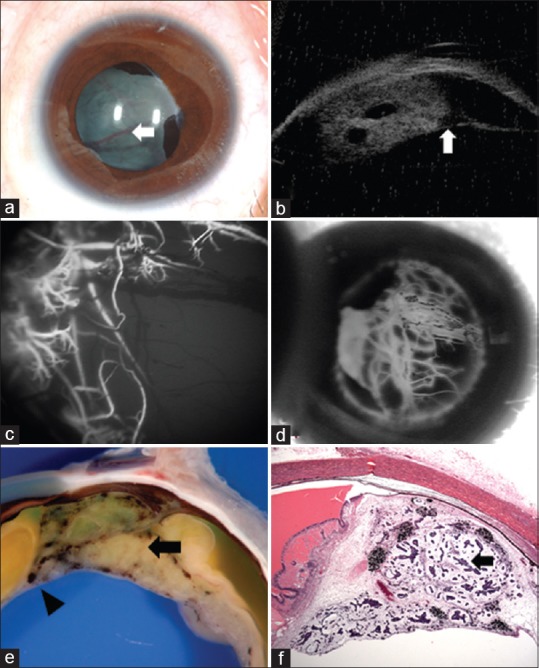
Intraocular medulloepithelioma is a nonhereditary neoplasm of childhood arising from primitive medullary epithelium. It most often involves the ciliary body. Most patients present between 2 and 10 years of age with loss of vision, pain, leucocoria, or conjunctival congestion. The mass appears as a grey-white ciliary body lesion with intratumoral cysts. Presence of a neoplastic cyclitic membrane with extension to retrolental region is characteristic. Secondary manifestations like cataract and neovascular glaucoma may be present in up to 50% and 60% patients, respectively. These could be the first signs for which, unfortunately, about 50% patients undergo surgery before recognition of the hidden tumor. Systemic correlation with pleuropulmonary blastoma (DICER1 gene) has been documented in 5% cases. Histopathology shows primitive neuroepithelial cells arranged as cords closely resembling the primitive retina. Histopathologically, the tumor is classified as teratoid (containing heteroplastic elements) and nonteratoid (containing medullary epithelial elements), each of which are further subclassified as benign or malignant. Retinoblastoma-like and sarcoma-like areas may be seen within the tissue. The treatment modality depends on tumor size and extent of invasion. For small localized tumors (≤3-4 clock hours), conservative treatments with cryotherapy, plaque radiotherapy, or partial lamellar sclerouvectomy (PLSU) have been used. Plaque brachytherapy is generally preferred for best tumor control. Advanced and extensive tumors require enucleation. Rare use of intra-arterial and intravitreal chemotherapy has been employed. Systemic prognosis is favorable, but those with extraocular extension and orbital involvement show risk for local recurrence and metastatic disease, which can lead to death.
眼内髓上皮瘤是一种儿童期非遗传性肿瘤,来源于原始髓上皮。它最常累及睫状体。大多数患者在 2 至 10 岁时出现视力丧失、疼痛、白瞳或结膜充血。肿瘤呈灰白色睫状体病变,伴有肿瘤内囊肿。肿瘤性睫状膜的存在及其向视网膜后区的延伸是特征性的。继发性表现如白内障和新生血管性青光眼分别可在多达 50%和 60%的患者中出现。这些可能是最初的迹象,不幸的是,大约 50%的患者在认识到隐藏的肿瘤之前就已经接受了手术。与胸膜肺胚细胞瘤(DICER1 基因)的系统相关性已在 5%的病例中得到证实。组织病理学显示原始神经上皮细胞排列成紧密类似于原始视网膜的索状。组织病理学上,肿瘤分为畸胎瘤(含有异型成分)和非畸胎瘤(含有髓上皮成分),两者进一步细分为良性或恶性。组织内可看到视网膜母细胞瘤样和肉瘤样区域。治疗方式取决于肿瘤的大小和侵袭程度。对于小的局限性肿瘤(≤3-4 时钟小时),可采用冷冻疗法、贴敷放疗或部分板层巩膜切除术(PLSU)进行保守治疗。贴敷近距离放疗通常更有利于肿瘤的控制。晚期和广泛的肿瘤需要眼球摘除。偶尔也会采用动脉内和玻璃体内化疗。全身预后良好,但那些有眼外侵犯和眼眶受累的患者有局部复发和转移疾病的风险,这可能导致死亡。