European Molecular Biology Laboratory, European Bioinformatics Institute (EMBL-EBI), Wellcome Trust Genome Campus, Cambridge, UK.
Bioinformatics. 2019 Nov 1;35(21):4493-4495. doi: 10.1093/bioinformatics/btz284.
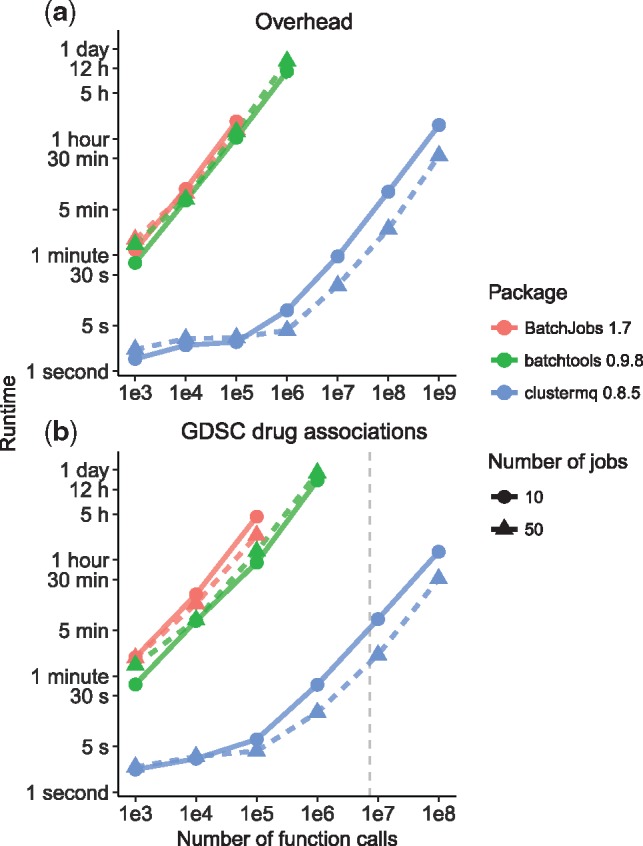
High performance computing (HPC) clusters play a pivotal role in large-scale bioinformatics analysis and modeling. For the statistical computing language R, packages exist to enable a user to submit their analyses as jobs on HPC schedulers. However, these packages do not scale well to high numbers of tasks, and their processing overhead quickly becomes a prohibitive bottleneck.
Here we present clustermq, an R package that can process analyses up to three orders of magnitude faster than previously published alternatives. We show this for investigating genomic associations of drug sensitivity in cancer cell lines, but it can be applied to any kind of parallelizable workflow.
The package is available on CRAN and https://github.com/mschubert/clustermq. Code for performance testing is available at https://github.com/mschubert/clustermq-performance.
Supplementary data are available at Bioinformatics online.
高性能计算(HPC)集群在大规模生物信息学分析和建模中起着至关重要的作用。对于统计计算语言 R,存在一些软件包可使用户将其分析作为 HPC 调度程序上的作业提交。但是,这些软件包并不能很好地扩展到大量任务,并且它们的处理开销很快成为一个难以逾越的瓶颈。
在这里,我们提出了 clustermq,这是一个 R 软件包,可以将分析速度提高到比以前发布的替代方案快三个数量级。我们以研究癌症细胞系中药物敏感性的基因组关联为例展示了这一点,但它可以应用于任何可并行化的工作流程。
该软件包可在 CRAN 和 https://github.com/mschubert/clustermq 上使用。性能测试的代码可在 https://github.com/mschubert/clustermq-performance 上获得。
补充数据可在生物信息学在线获得。