Department of Biochemistry & Molecular Biology, School of Neurobiology, Biochemistry & Biophysics, Life Sciences Faculty, Tel Aviv University, Tel Aviv 69978, Israel.
Signaling Systems Section, Laboratory of Immune System Biology, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, 20892 MD, USA.
Mediators Inflamm. 2019 Apr 17;2019:3451461. doi: 10.1155/2019/3451461. eCollection 2019.
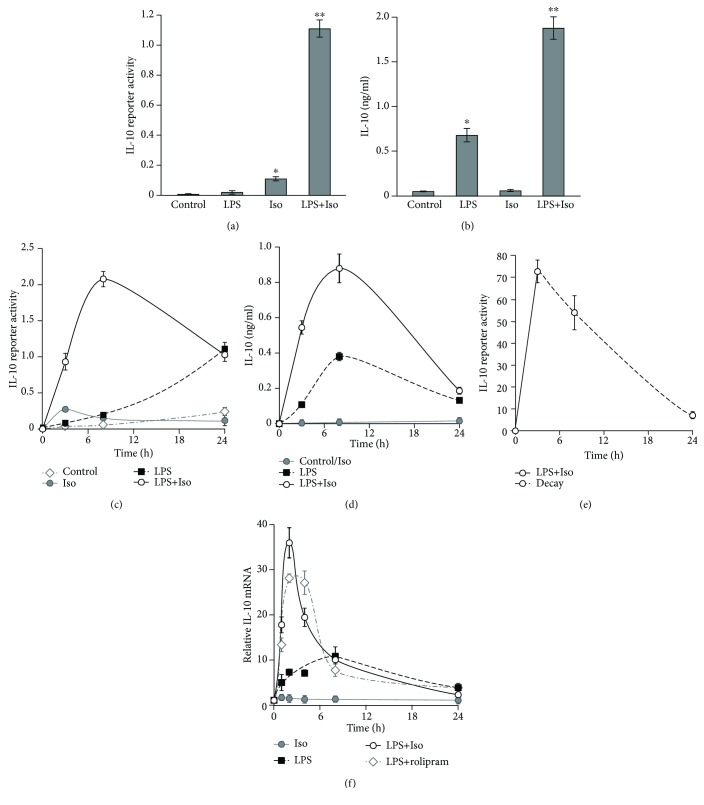
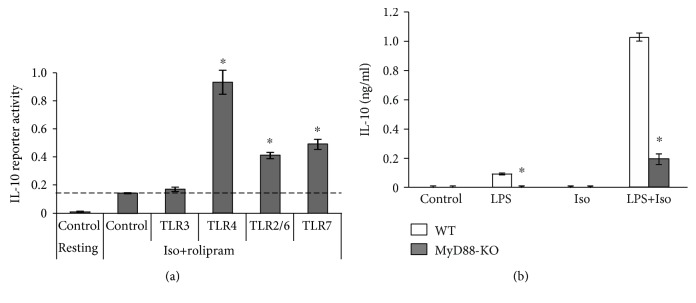
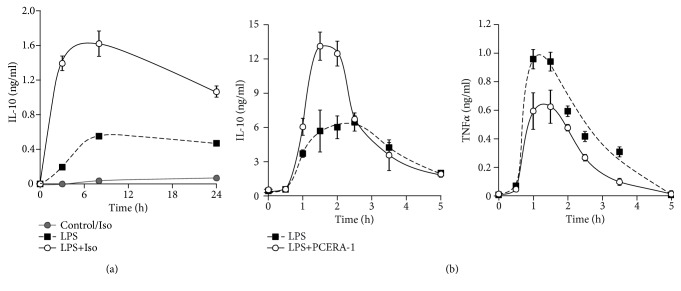
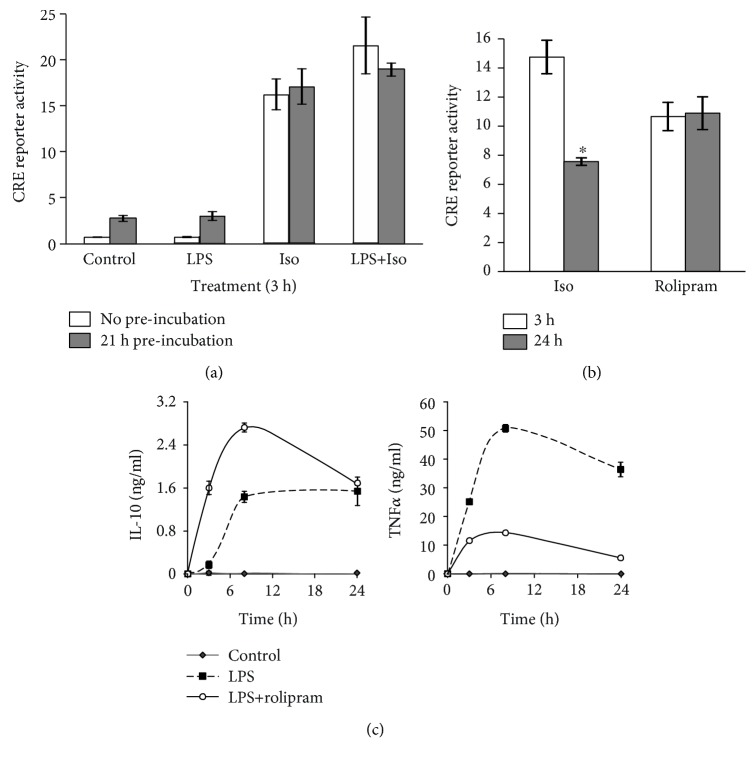
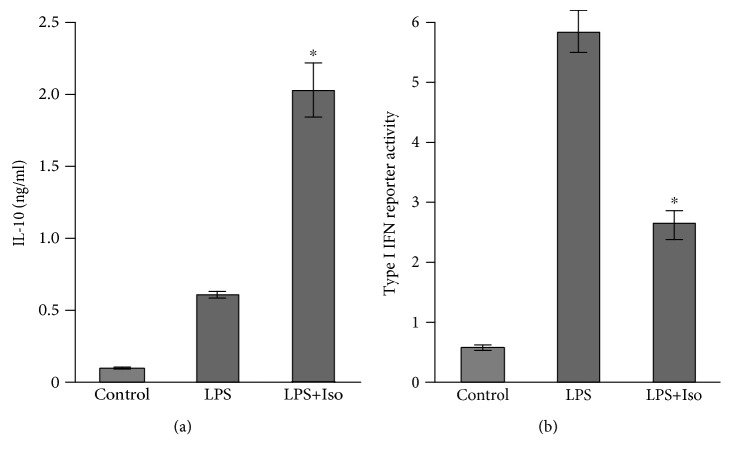
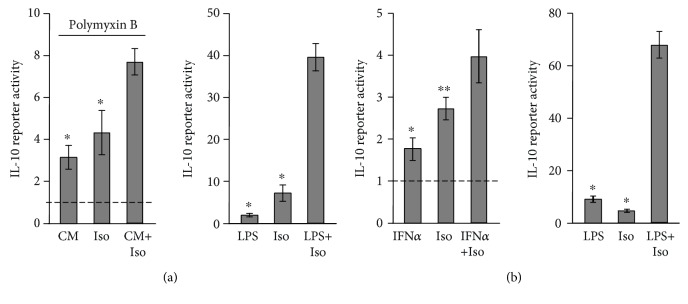
Interleukin-10 (IL-10) is a key anti-inflammatory cytokine, secreted by macrophages and other immune cells to attenuate inflammation. Autocrine type I interferons (IFNs) largely mediate the delayed expression of IL-10 by LPS-stimulated macrophages. We have previously shown that IL-10 is synergistically expressed in macrophages following a costimulus of a TLR agonist and cAMP. We now show that the cAMP pathway directly upregulates IL-10 transcription and plays an important permissive and synergistic role in early, but not late, LPS-stimulated IL-10 mRNA and protein expression in mouse macrophages and in a mouse septic shock model. Our results suggest that the loss of synergism is not due to desensitization of the cAMP inducing signal, and it is not mediated by a positive crosstalk between the cAMP and type I IFN pathways. First, cAMP elevation in LPS-treated cells decreased the secretion of type I IFN. Second, autocrine/paracrine type I IFNs induce IL-10 promoter reporter activity only additively, but not synergistically, with the cAMP pathway. IL-10 promoter reporter activity was synergistically induced by cAMP elevation in macrophages stimulated by an agonist of either TLR4, TLR2/6, or TLR7, receptors which signal via MyD88, but not by an agonist of TLR3 which signals independently of MyD88. Moreover, MyD88 knockout largely reduced the synergistic IL-10 expression, indicating that MyD88 is required for the synergism displayed by LPS with cAMP. This report delineates the temporal regulation of early cAMP-accelerated vs. late type I IFN-dependent IL-10 transcription in LPS-stimulated murine macrophages that can limit inflammation at its onset.
白细胞介素-10(IL-10)是一种关键的抗炎细胞因子,由巨噬细胞和其他免疫细胞分泌,以减弱炎症反应。自分泌 I 型干扰素(IFN)在很大程度上介导 LPS 刺激的巨噬细胞中 IL-10 的延迟表达。我们之前已经表明,在 TLR 激动剂和 cAMP 的共刺激下,巨噬细胞中协同表达 IL-10。我们现在表明,cAMP 途径直接上调 IL-10 转录,并在早期(但不是晚期)LPS 刺激的小鼠巨噬细胞和小鼠脓毒症休克模型中对 IL-10 mRNA 和蛋白质表达发挥重要的许可和协同作用。我们的结果表明,协同作用的丧失不是由于 cAMP 诱导信号的脱敏,也不是由 cAMP 和 I 型 IFN 途径之间的正交叉对话介导的。首先,LPS 处理细胞中的 cAMP 升高会降低 I 型 IFN 的分泌。其次,自分泌/旁分泌 I 型 IFNs 仅以累加方式而不是协同方式诱导 IL-10 启动子报告基因活性与 cAMP 途径。cAMP 升高在 TLR4、TLR2/6 或 TLR7 激动剂刺激的巨噬细胞中协同诱导 IL-10 启动子报告基因活性,而 TLR3 激动剂(独立于 MyD88 信号)则不协同诱导。此外,MyD88 敲除大大降低了协同的 IL-10 表达,表明 MyD88 是 LPS 与 cAMP 显示协同作用所必需的。本报告描绘了 LPS 刺激的小鼠巨噬细胞中早期 cAMP 加速与晚期 I 型 IFN 依赖性 IL-10 转录的时间调节,这可以限制炎症的发生。