Babarinde Isaac A, Li Yuhao, Hutchins Andrew P
Department of Biology, Southern University of Science and Technology, 1088 Xueyuan Lu, Shenzhen, China.
Comput Struct Biotechnol J. 2019 May 7;17:628-637. doi: 10.1016/j.csbj.2019.04.012. eCollection 2019.
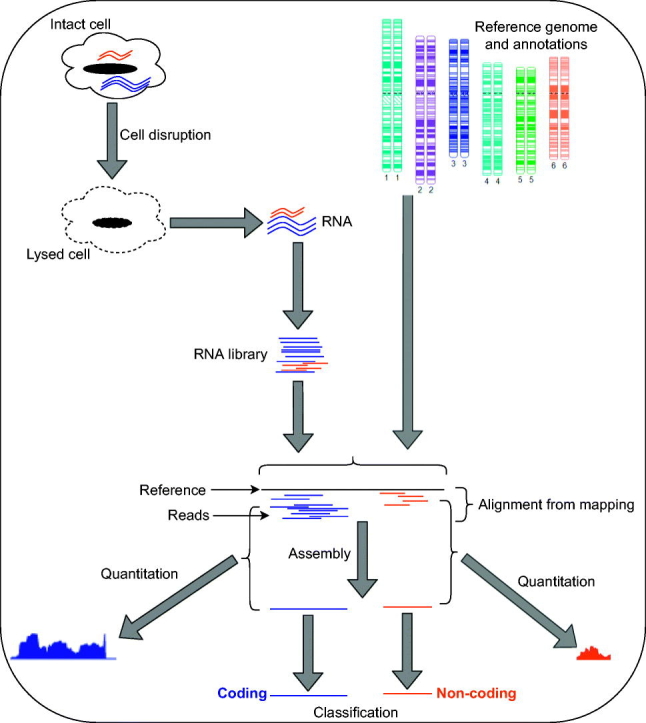
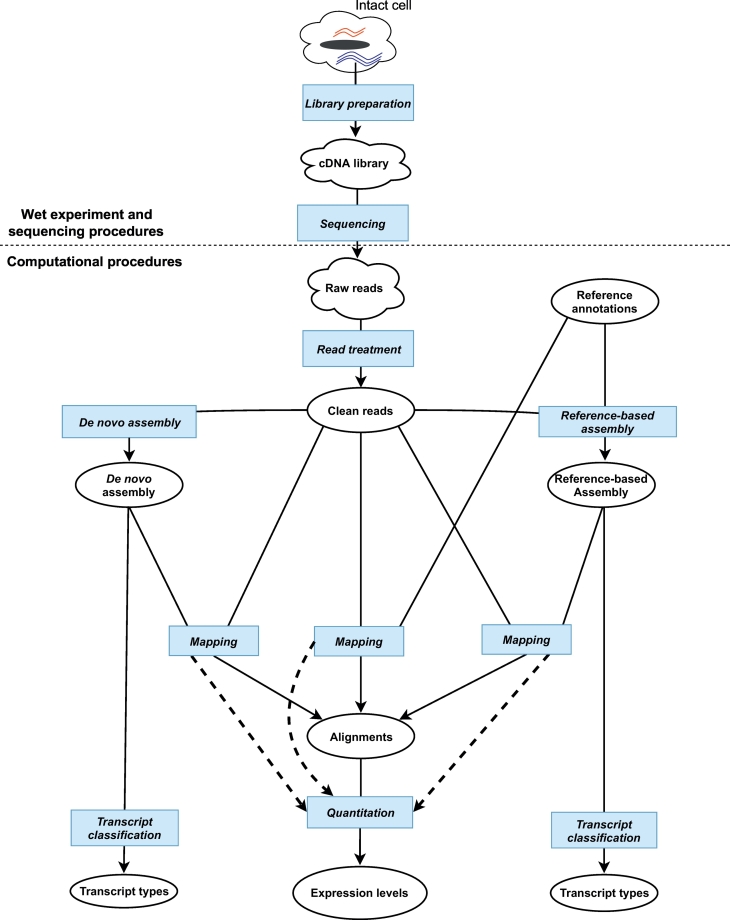
The measurement of gene expression has long provided significant insight into biological functions. The development of high-throughput short-read sequencing technology has revealed transcriptional complexity at an unprecedented scale, and informed almost all areas of biology. However, as researchers have sought to gather more insights from the data, these new technologies have also increased the computational analysis burden. In this review, we describe typical computational pipelines for RNA-Seq analysis and discuss their strengths and weaknesses for the assembly, quantification and analysis of coding and non-coding RNAs. We also discuss the assembly of transposable elements into transcripts, and the difficulty these repetitive elements pose. In summary, RNA-Seq is a powerful technology that is likely to remain a key asset in the biologist's toolkit.
长期以来,基因表达的测量为深入了解生物学功能提供了重要依据。高通量短读长测序技术的发展以前所未有的规模揭示了转录复杂性,并为几乎所有生物学领域提供了信息。然而,随着研究人员试图从数据中获取更多见解,这些新技术也增加了计算分析负担。在本综述中,我们描述了RNA测序分析的典型计算流程,并讨论了它们在编码和非编码RNA的组装、定量和分析方面的优缺点。我们还讨论了将转座元件组装成转录本的问题,以及这些重复元件带来的困难。总之,RNA测序是一项强大的技术,很可能仍然是生物学家工具包中的关键资产。