Key Laboratory of Biology and Genetic Improvement of Maize in Southwest Region, Maize Research Institute, Sichuan Agricultural University, Chengdu, China.
Department of Agronomy, Iowa State University, Ames, IA, USA.
Plant Biotechnol J. 2020 Jan;18(1):207-221. doi: 10.1111/pbi.13188. Epub 2019 Jun 26.
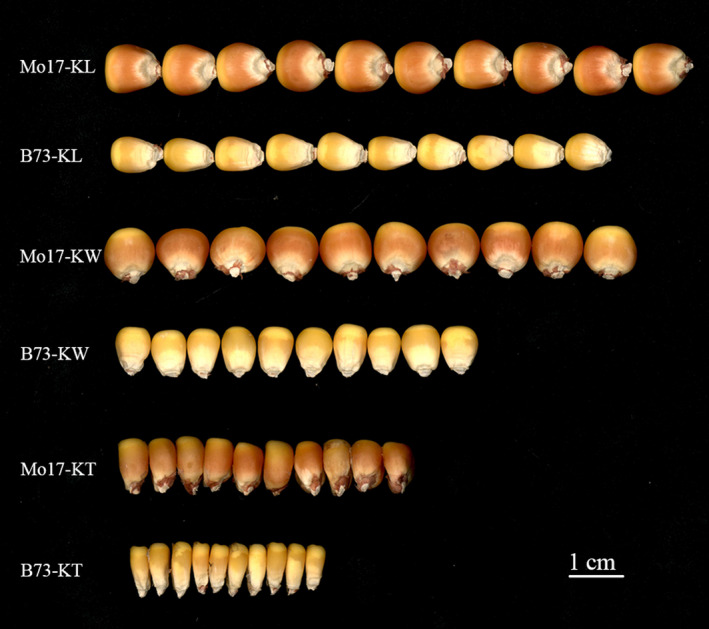
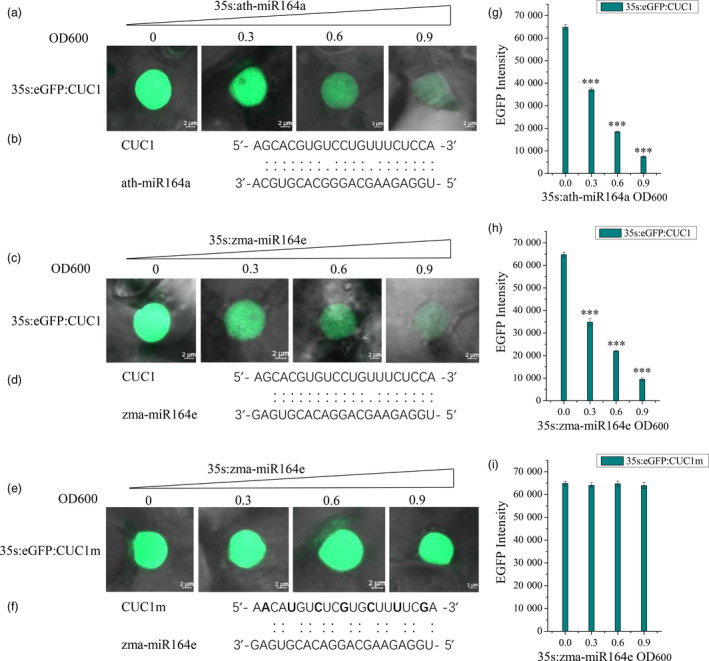
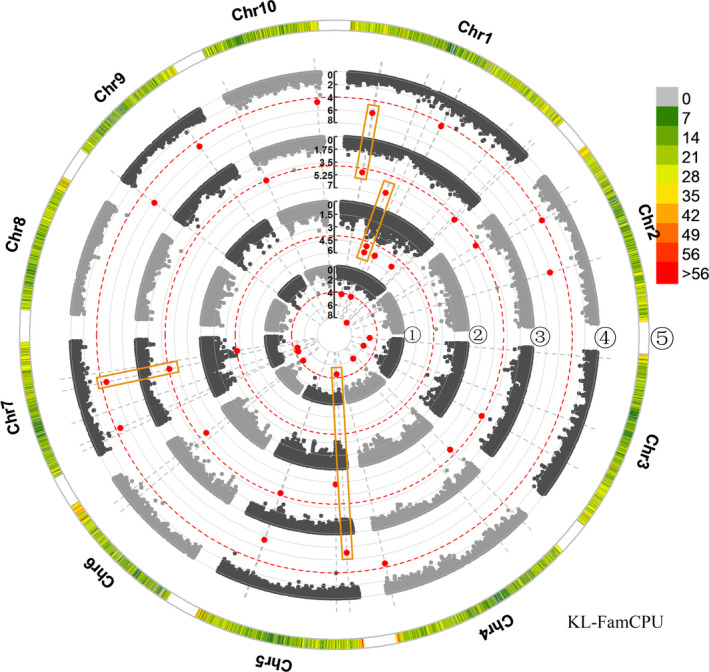
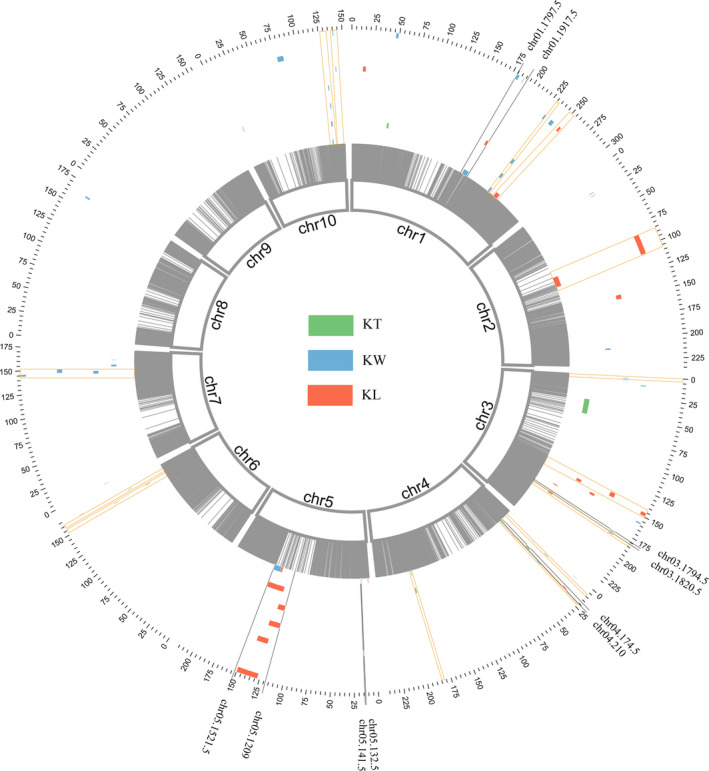
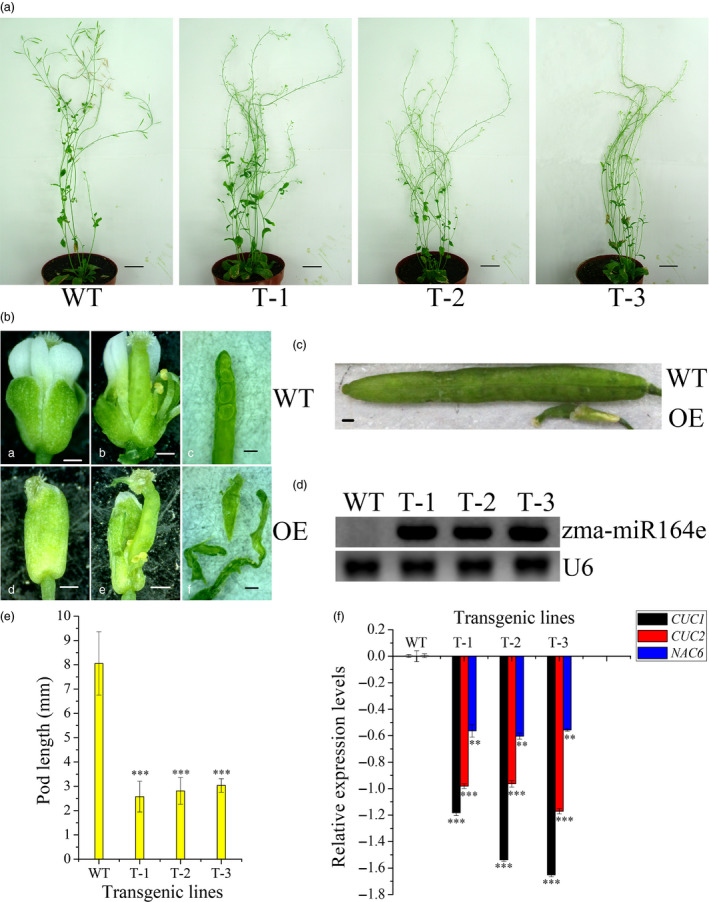
Kernel size-related traits are the most direct traits correlating with grain yield. The genetic basis of three kernel traits of maize, kernel length (KL), kernel width (KW) and kernel thickness (KT), was investigated in an association panel and a biparental population. A total of 21 single nucleotide polymorphisms (SNPs) were detected to be most significantly (P < 2.25 × 10 ) associated with these three traits in the association panel under four environments. Furthermore, 50 quantitative trait loci (QTL) controlling these traits were detected in seven environments in the intermated B73 × Mo17 (IBM) Syn10 doubled haploid (DH) population, of which eight were repetitively identified in at least three environments. Combining the two mapping populations revealed that 56 SNPs (P < 1 × 10 ) fell within 18 of the QTL confidence intervals. According to the top significant SNPs, stable-effect SNPs and the co-localized SNPs by association analysis and linkage mapping, a total of 73 candidate genes were identified, regulating seed development. Additionally, seven miRNAs were found to situate within the linkage disequilibrium (LD) regions of the co-localized SNPs, of which zma-miR164e was demonstrated to cleave the mRNAs of Arabidopsis CUC1, CUC2 and NAC6 in vitro. Overexpression of zma-miR164e resulted in the down-regulation of these genes above and the failure of seed formation in Arabidopsis pods, with the increased branch number. These findings provide insights into the mechanism of seed development and the improvement of molecular marker-assisted selection (MAS) for high-yield breeding in maize.
与粒宽相关的性状是与粒重最直接相关的性状。本研究利用关联群体和双交群体分析了玉米三个粒宽相关性状(粒长(KL)、粒宽(KW)和粒厚(KT))的遗传基础。在四个环境下,关联群体中共有 21 个单核苷酸多态性(SNP)被检测到与这三个性状显著关联(P < 2.25×10)。此外,在互交 B73×Mo17(IBM)Syn10 加倍单倍体(DH)群体的七个环境中,检测到 50 个控制这些性状的数量性状位点(QTL),其中 8 个在至少三个环境中重复检测到。将两个作图群体相结合,发现 56 个 SNP(P < 1×10)位于 18 个 QTL 置信区间内。根据关联分析和连锁作图的最高显著 SNP、稳定效应 SNP 和共定位 SNP,共鉴定到 73 个候选基因,它们参与种子发育的调控。此外,在连锁不平衡(LD)区域内还发现了七个 miRNA,其中 zma-miR164e 被证明可以体外切割拟南芥 CUC1、CUC2 和 NAC6 的 mRNA。过表达 zma-miR164e 导致这些基因的下调以及拟南芥蒴果中种子形成失败,同时分支数量增加。这些发现为种子发育机制提供了新的认识,并为玉米高产分子标记辅助选择(MAS)改良提供了理论依据。