Department of Prosthetic Dentistry and Biomedical Materials Science, Hannover Medical School (MHH), Hannover, Germany.
Lower Saxony Centre for Biomedical Engineering, Implant Research and Development (NIFE), Hannover, Germany.
ISME J. 2019 Oct;13(10):2500-2522. doi: 10.1038/s41396-019-0450-8. Epub 2019 Jun 14.
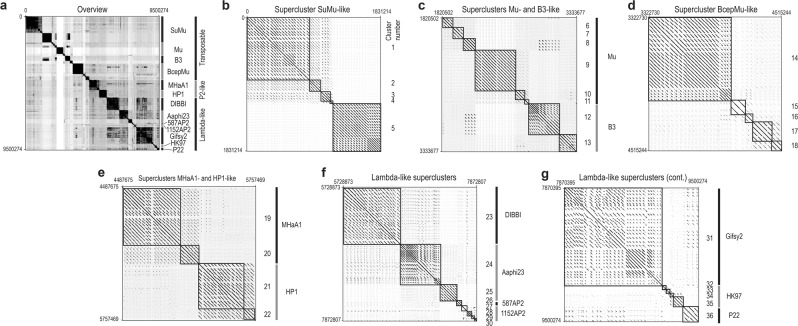
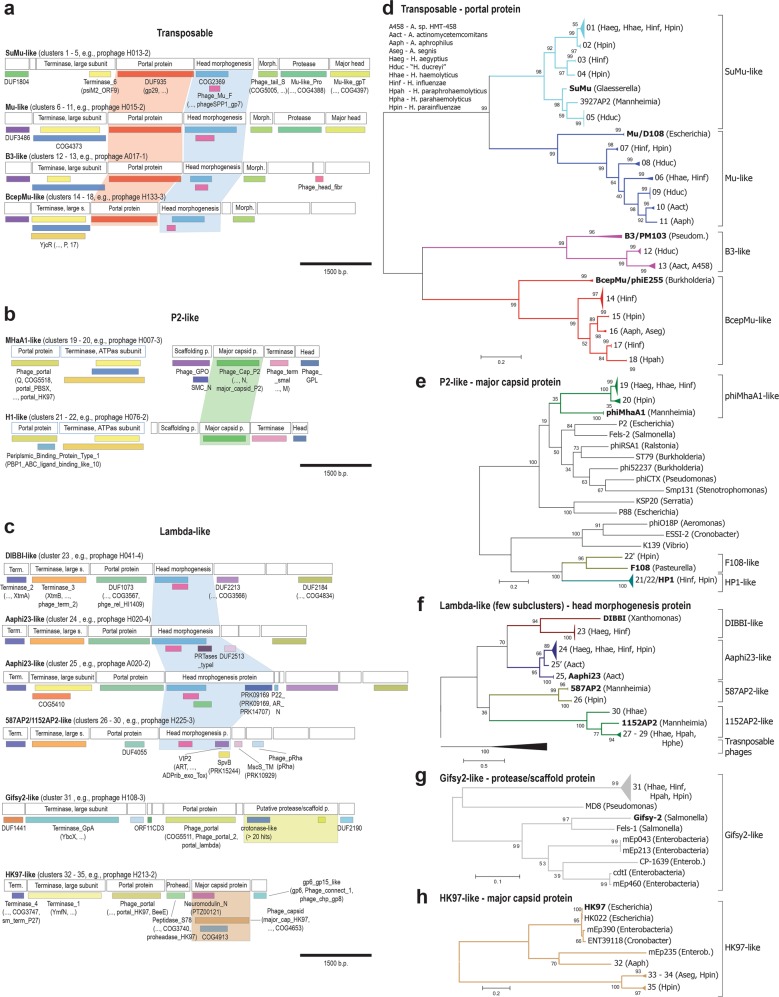
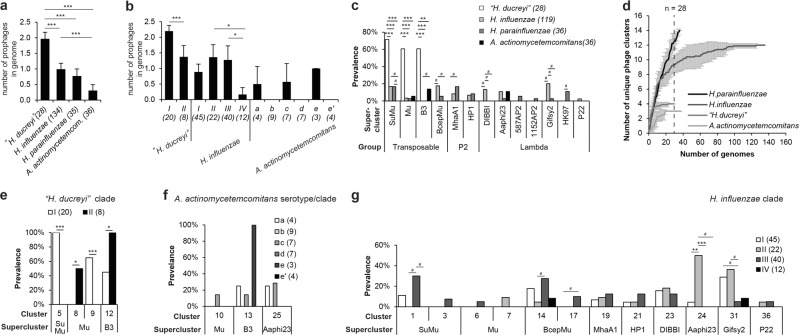
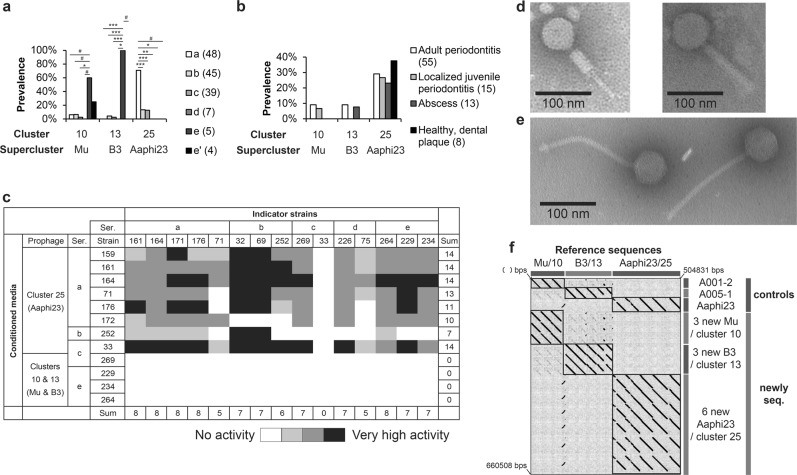
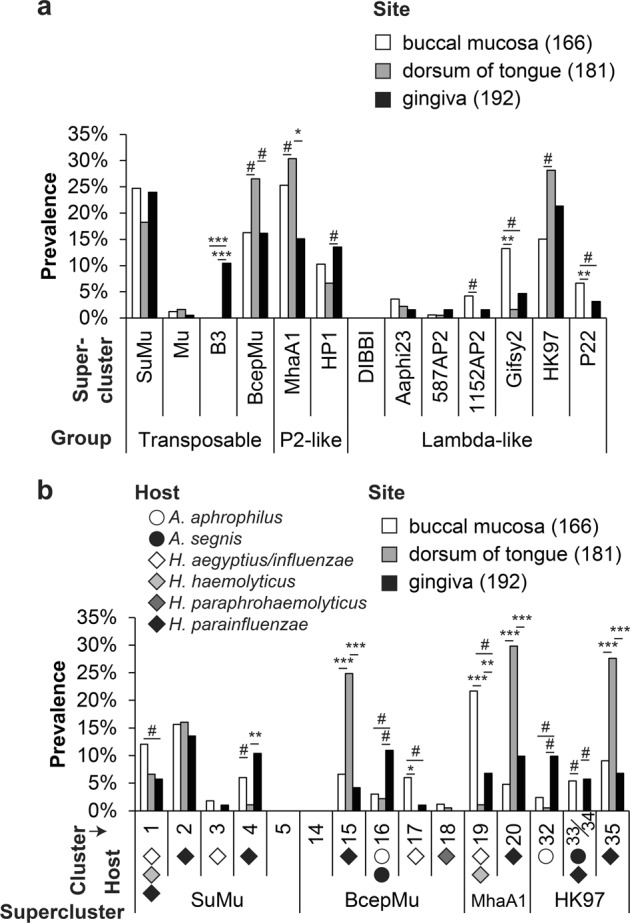
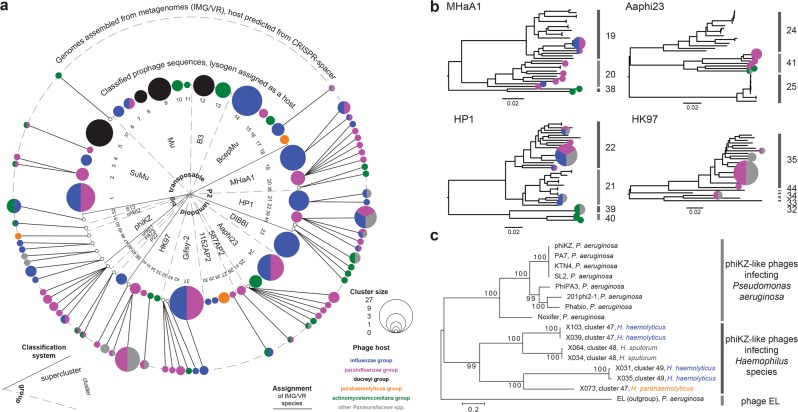
Aggregatibacter and Haemophilus species are relevant human commensals and opportunistic pathogens. Consequently, their bacteriophages may have significant impact on human microbial ecology and pathologies. Our aim was to reveal the prevalence and diversity of bacteriophages infecting Aggregatibacter and Haemophilus species that colonize the human body. Genome mining with comparative genomics, screening of clinical isolates, and profiling of metagenomes allowed characterization of 346 phages grouped in 52 clusters and 18 superclusters. Less than 10% of the identified phage clusters were represented by previously characterized phages. Prophage diversity patterns varied significantly for different phage types, host clades, and environmental niches. A more diverse phage community lysogenizes Haemophilus influenzae and Haemophilus parainfluenzae strains than Aggregatibacter actinomycetemcomitans and "Haemophilus ducreyi". Co-infections occurred more often in "H. ducreyi". Phages from Aggregatibacter actinomycetemcomitans preferably lysogenized strains of specific serotype. Prophage patterns shared by subspecies clades of different bacterial species suggest similar ecoevolutionary drivers. Changes in frequencies of DNA uptake signal sequences and guanine-cytosine content reflect phage-host long-term coevolution. Aggregatibacter and Haemophilus phages were prevalent at multiple oral sites. Together, these findings should help exploring the ecoevolutionary forces shaping virus-host interactions in the human microbiome. Putative lytic phages, especially phiKZ-like, may provide new therapeutic options.
聚集菌属和嗜血菌属是相关的人体共生菌和机会性病原体。因此,它们的噬菌体可能对人类微生物生态和病理学有重大影响。我们的目的是揭示感染定植于人体的聚集菌属和嗜血菌属的噬菌体的流行情况和多样性。通过比较基因组学进行基因组挖掘、筛选临床分离株以及对宏基因组进行分析,我们鉴定了 346 种噬菌体,它们分为 52 个聚类和 18 个超级聚类。所鉴定的噬菌体聚类中,不到 10%的聚类由以前表征的噬菌体代表。不同的噬菌体类型、宿主进化枝和环境生态位的噬菌体多样性模式差异显著。与Aggregatibacter actinomycetemcomitans 和“嗜血流感菌”相比,更多样化的噬菌体群落可溶菌感染流感嗜血杆菌和副流感嗜血杆菌。“嗜血流感菌”更容易发生合并感染。来自 Aggregatibacter actinomycetemcomitans 的噬菌体更喜欢溶菌特定血清型的菌株。不同细菌物种亚种进化枝之间共享的噬菌体模式表明存在类似的生态进化驱动力。DNA 摄取信号序列和鸟嘌呤-胞嘧啶含量的变化反映了噬菌体-宿主的长期共同进化。Aggregatibacter 和嗜血菌属的噬菌体在多个口腔部位普遍存在。这些发现共同有助于探索塑造人类微生物组中病毒-宿主相互作用的生态进化力量。潜在的裂解噬菌体,特别是 phiKZ 样噬菌体,可能提供新的治疗选择。