Li Xiaoqian, Liu Zijian, Mi Mi, Zhang Caijiao, Xiao Yin, Liu Xinxiu, Wu Gang, Zhang Liling
Cancer Center, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, People's Republic of China.
Cancer Manag Res. 2019 Jun 6;11:5209-5220. doi: 10.2147/CMAR.S185030. eCollection 2019.
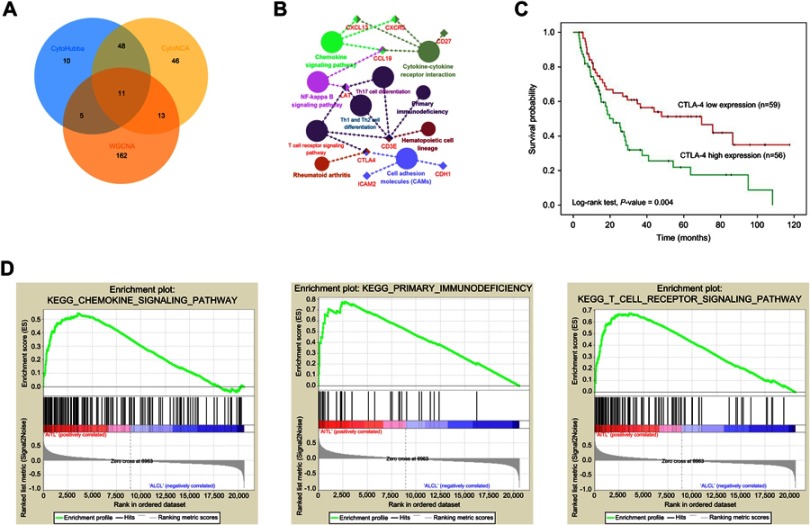
Angioimmunoblastic T-cell lymphoma (AITL) is an aggressive subtype of peripheral T-cell lymphoma (PTCL) that has a poor 5-year overall survival rate due to its lack of precise therapeutic targets. Identifying potential prognostic markers of AITL may provide information regarding the development of precision medicine. RNA sequence data from PTCL and patient clinic traits were obtained from the Gene Expression Omnibus (GEO) database. Differentially expressed gene (DEG) analysis and weighted gene co-expression network analysis (WGCNA) were performed to identify DEGs between the different PTCL subtypes and investigate the relationship underlying co-expression modules and clinic traits. Gene ontology (GO) and protein-protein interaction (PPI) network analyses based on DAVID and the STRING website, respectively, were utilized to deeply excavate hub genes. After removing the outliers from the GSE65823, GSE58445, GSE19069, and GSE6338 datasets using the results from an unsupervised cluster heatmap, 50 AITL samples and 55 anaplastic large cell lymphoma (ALCL) samples were screened. A total of 677 upregulated DEGs and 237 downregulated DEGs were identified in AITL and used to construct a PPI network complex. Using WGCNA, 12 identified co-expression modules were constructed from the 5468 genes with the top 10% of variance, and 192 genes from the Turquoise and Brown modules were with a Gene Significance (GS) cut-off threshold >0.6. Eleven hub genes (CDH1, LAT, LPAR1, CXCL13, CD27, ICAM2, CD3E, CCL19, CTLA-4, CXCR5, and C3) were identified. Only CTLA-4 overexpressed was found to be a poor prognostic factor according to survival analysis. Gene set enrichment analysis (GSEA) identified and validated the intersection of key pathways (T cell receptor, primary immunodeficiency, and chemokine signaling pathways). Our findings provide the framework for the identification of AITL co-expression gene modules and identify key pathways and driving genes that may be novel treatment targets and helpful for the development of a prognostic evaluation index.
血管免疫母细胞性T细胞淋巴瘤(AITL)是外周T细胞淋巴瘤(PTCL)的一种侵袭性亚型,由于缺乏精确的治疗靶点,其5年总生存率较低。确定AITL的潜在预后标志物可能为精准医学的发展提供信息。从基因表达综合数据库(GEO)获取PTCL的RNA序列数据和患者临床特征。进行差异表达基因(DEG)分析和加权基因共表达网络分析(WGCNA),以识别不同PTCL亚型之间的DEG,并研究共表达模块与临床特征之间的潜在关系。分别基于DAVID和STRING网站进行基因本体(GO)和蛋白质-蛋白质相互作用(PPI)网络分析,以深入挖掘核心基因。使用无监督聚类热图的结果从GSE65823、GSE58445、GSE19069和GSE6338数据集中去除异常值后,筛选出50个AITL样本和55个间变性大细胞淋巴瘤(ALCL)样本。在AITL中总共鉴定出677个上调的DEG和237个下调的DEG,并用于构建PPI网络复合体。使用WGCNA,从方差前10%的5468个基因中构建了12个鉴定出的共表达模块,来自绿松石和棕色模块的192个基因的基因显著性(GS)截止阈值>0.6。鉴定出11个核心基因(CDH1、LAT、LPAR1、CXCL13、CD27、ICAM2、CD3E、CCL19、CTLA-4、CXCR5和C3)。根据生存分析,仅发现CTLA-4过表达是一个不良预后因素。基因集富集分析(GSEA)鉴定并验证了关键通路(T细胞受体、原发性免疫缺陷和趋化因子信号通路)的交集。我们的研究结果为鉴定AITL共表达基因模块提供了框架,并确定了可能成为新治疗靶点的关键通路和驱动基因,有助于开发预后评估指标。