Brouard Jean-Simon, Schenkel Flavio, Marete Andrew, Bissonnette Nathalie
1Sherbrooke Research and Development Centre, Agriculture and Agri-Food Canada, Sherbrooke, QC J1M 0C8 Canada.
2Center of Genetic Improvement of Livestock, University of Guelph, Guelph, ON N1G 2W1 Canada.
J Anim Sci Biotechnol. 2019 Jun 21;10:44. doi: 10.1186/s40104-019-0359-0. eCollection 2019.
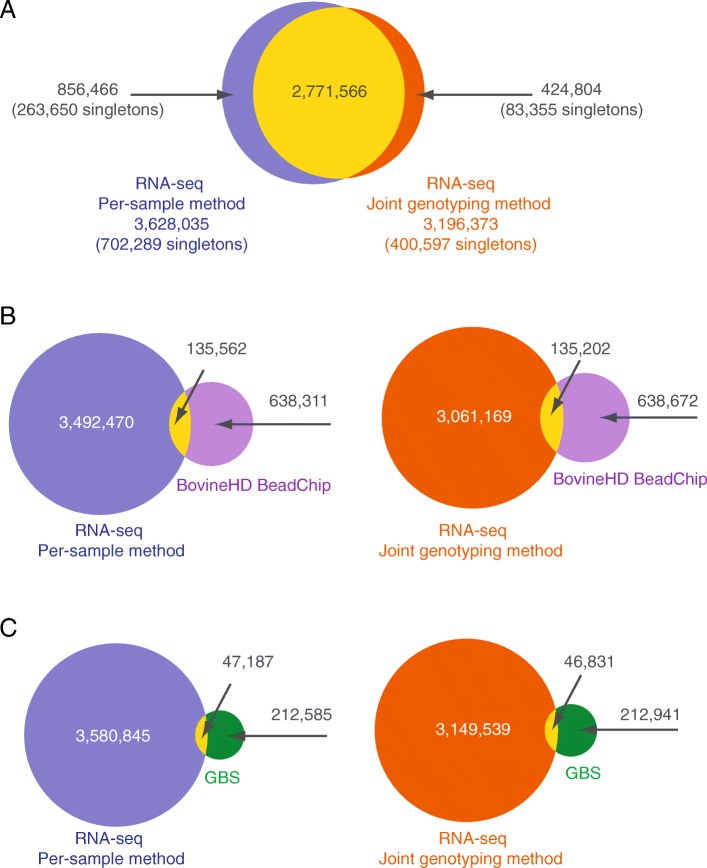
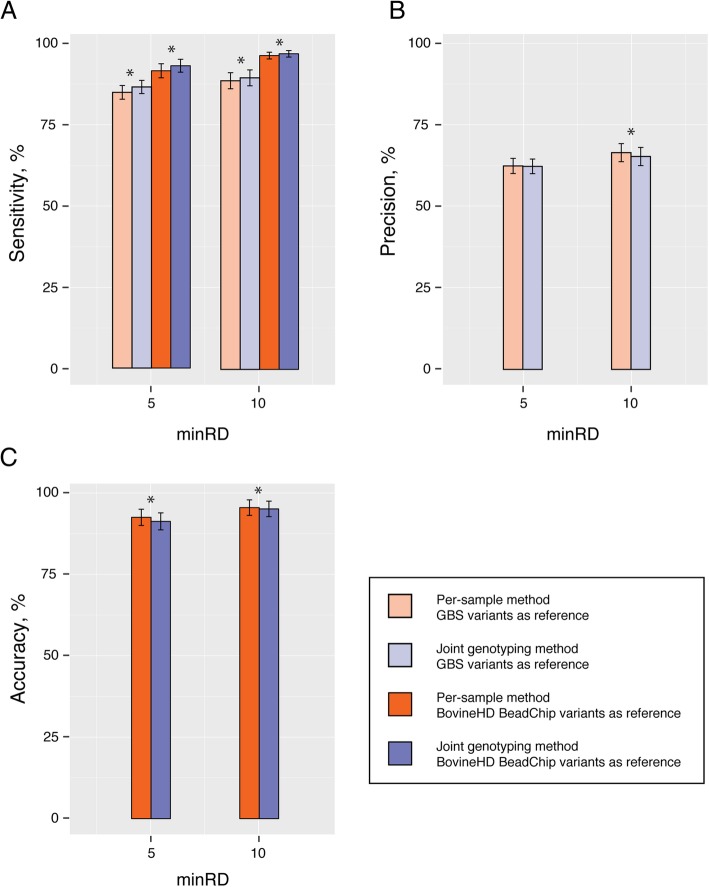
The Genome Analysis Toolkit (GATK) is a popular set of programs for discovering and genotyping variants from next-generation sequencing data. The current GATK recommendation for RNA sequencing (RNA-seq) is to perform variant calling from individual samples, with the drawback that only variable positions are reported. Versions 3.0 and above of GATK offer the possibility of calling DNA variants on cohorts of samples using the HaplotypeCaller algorithm in Genomic Variant Call Format (GVCF) mode. Using this approach, variants are called individually on each sample, generating one GVCF file per sample that lists genotype likelihoods and their genome annotations. In a second step, variants are called from the GVCF files through a joint genotyping analysis. This strategy is more flexible and reduces computational challenges in comparison to the traditional joint discovery workflow. Using a GVCF workflow for mining SNP in RNA-seq data provides substantial advantages, including reporting homozygous genotypes for the reference allele as well as missing data. Taking advantage of RNA-seq data derived from primary macrophages isolated from 50 cows, the GATK joint genotyping method for calling variants on RNA-seq data was validated by comparing this approach to a so-called "per-sample" method. In addition, pair-wise comparisons of the two methods were performed to evaluate their respective sensitivity, precision and accuracy using DNA genotypes from a companion study including the same 50 cows genotyped using either genotyping-by-sequencing or with the Bovine SNP50 Beadchip (imputed to the Bovine high density). Results indicate that both approaches are very close in their capacity of detecting reference variants and that the joint genotyping method is more sensitive than the per-sample method. Given that the joint genotyping method is more flexible and technically easier, we recommend this approach for variant calling in RNA-seq experiments.
基因组分析工具包(GATK)是一套用于从下一代测序数据中发现和基因分型变异的常用程序。目前GATK对RNA测序(RNA-seq)的建议是对单个样本进行变异检测,缺点是只报告可变位置。GATK 3.0及以上版本提供了在基因组变异调用格式(GVCF)模式下使用单倍型分型算法对样本队列进行DNA变异检测的可能性。使用这种方法,对每个样本单独进行变异检测,每个样本生成一个GVCF文件,列出基因型似然性及其基因组注释。第二步,通过联合基因分型分析从GVCF文件中检测变异。与传统的联合发现工作流程相比,这种策略更灵活,减少了计算挑战。使用GVCF工作流程挖掘RNA-seq数据中的单核苷酸多态性(SNP)具有显著优势,包括报告参考等位基因的纯合基因型以及缺失数据。利用从50头奶牛分离的原代巨噬细胞获得的RNA-seq数据,通过将该方法与所谓的“单样本”方法进行比较,验证了GATK联合基因分型方法在RNA-seq数据上检测变异的能力。此外,使用来自一项配套研究的DNA基因型对这两种方法进行成对比较,该研究对包括这50头奶牛在内的样本分别使用测序分型或牛SNP50芯片(推算为牛高密度芯片)进行基因分型,以评估它们各自的灵敏度、精密度和准确性。结果表明,两种方法在检测参考变异的能力上非常接近,联合基因分型方法比单样本方法更灵敏。鉴于联合基因分型方法更灵活且技术上更简便,我们建议在RNA-seq实验中使用这种方法进行变异检测。