Oikkonen Laura, Lise Stefano
Wellcome Trust Centre for Human Genetics, University of Oxford, Oxford, UK.
Centre for Evolution and Cancer, The Institute of Cancer Research, Sutton, UK.
Wellcome Open Res. 2017 Jan 17;2:6. doi: 10.12688/wellcomeopenres.10501.2.
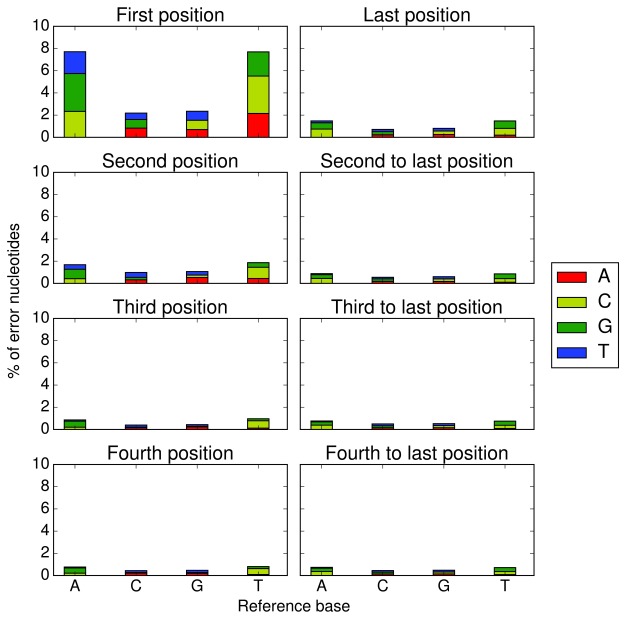
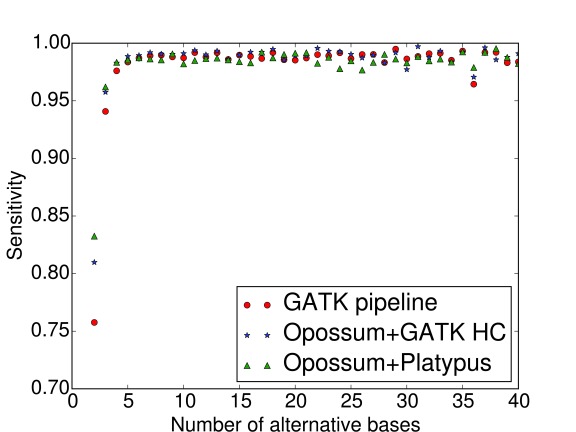
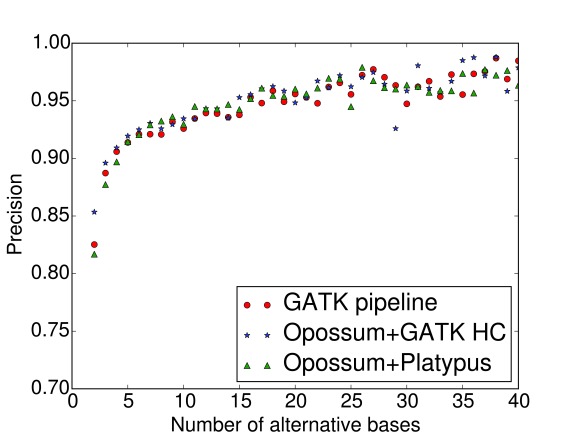
Identifying variants from RNA-seq (transcriptome sequencing) data is a cost-effective and versatile alternative to whole-genome sequencing. However, current variant callers do not generally behave well with RNA-seq data due to reads encompassing intronic regions. We have developed a software programme called Opossum to address this problem. Opossum pre-processes RNA-seq reads prior to variant calling, and although it has been designed to work specifically with Platypus, it can be used equally well with other variant callers such as GATK HaplotypeCaller. In this work, we show that using Opossum in conjunction with either Platypus or GATK HaplotypeCaller maintains precision and improves the sensitivity for SNP detection compared to the GATK Best Practices pipeline. In addition, using it in combination with Platypus offers a substantial reduction in run times compared to the GATK pipeline so it is ideal when there are only limited time or computational resources available.
从RNA测序(转录组测序)数据中识别变异体是一种经济高效且通用的全基因组测序替代方法。然而,由于读取的序列包含内含子区域,当前的变异体调用程序在处理RNA测序数据时通常表现不佳。我们开发了一个名为负鼠(Opossum)的软件程序来解决这个问题。负鼠在进行变异体调用之前会对RNA测序读取的数据进行预处理,虽然它是专门为与鸭嘴兽(Platypus)一起使用而设计的,但它也可以与其他变异体调用程序(如GATK HaplotypeCaller)同样出色地配合使用。在这项工作中,我们表明,与GATK最佳实践流程相比,将负鼠与鸭嘴兽或GATK HaplotypeCaller结合使用可保持精度并提高单核苷酸多态性(SNP)检测的灵敏度。此外,与GATK流程相比,将其与鸭嘴兽结合使用可大幅减少运行时间,因此在时间或计算资源有限时非常理想。