Centre for Discovery Brain Sciences, Edinburgh Medical School: Biomedical Sciences, Edinburgh, UK.
Euan MacDonald Centre for Motor Neurone Disease Research, University of Edinburgh, Edinburgh, Scotland, EH8 9XD, UK.
Cell Death Dis. 2019 Jul 4;10(7):515. doi: 10.1038/s41419-019-1727-6.
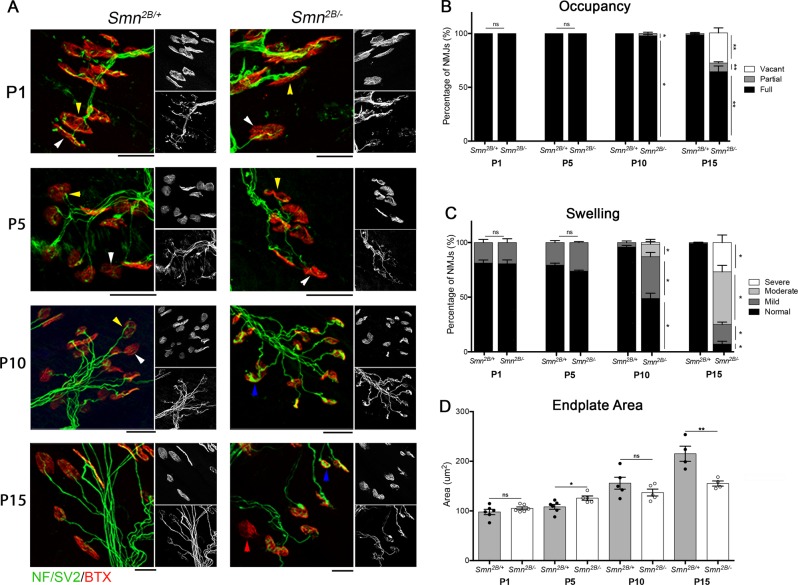
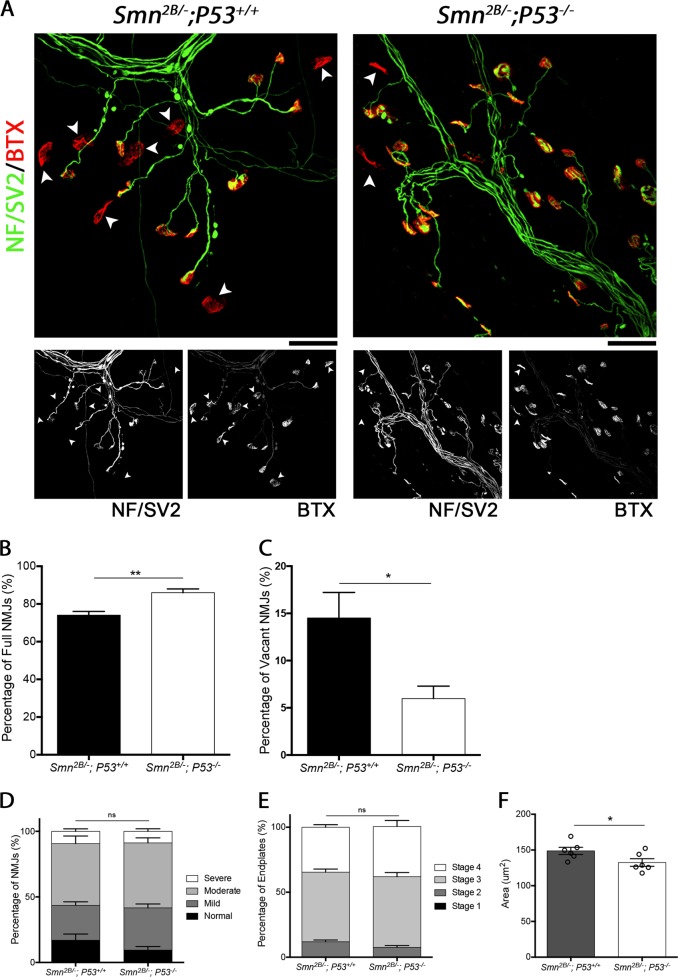
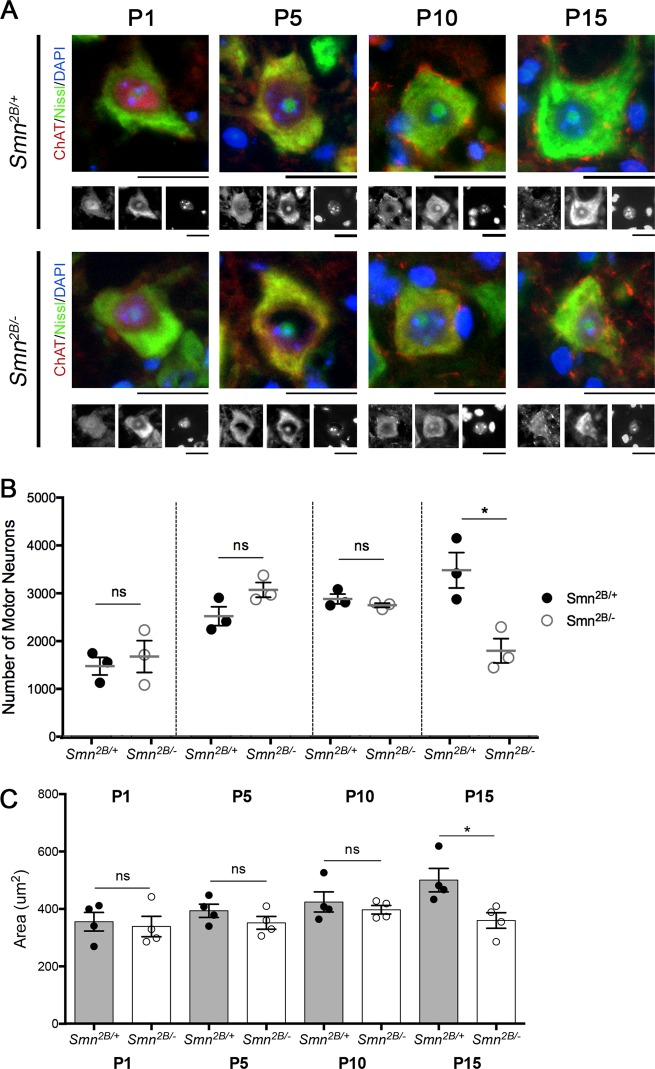
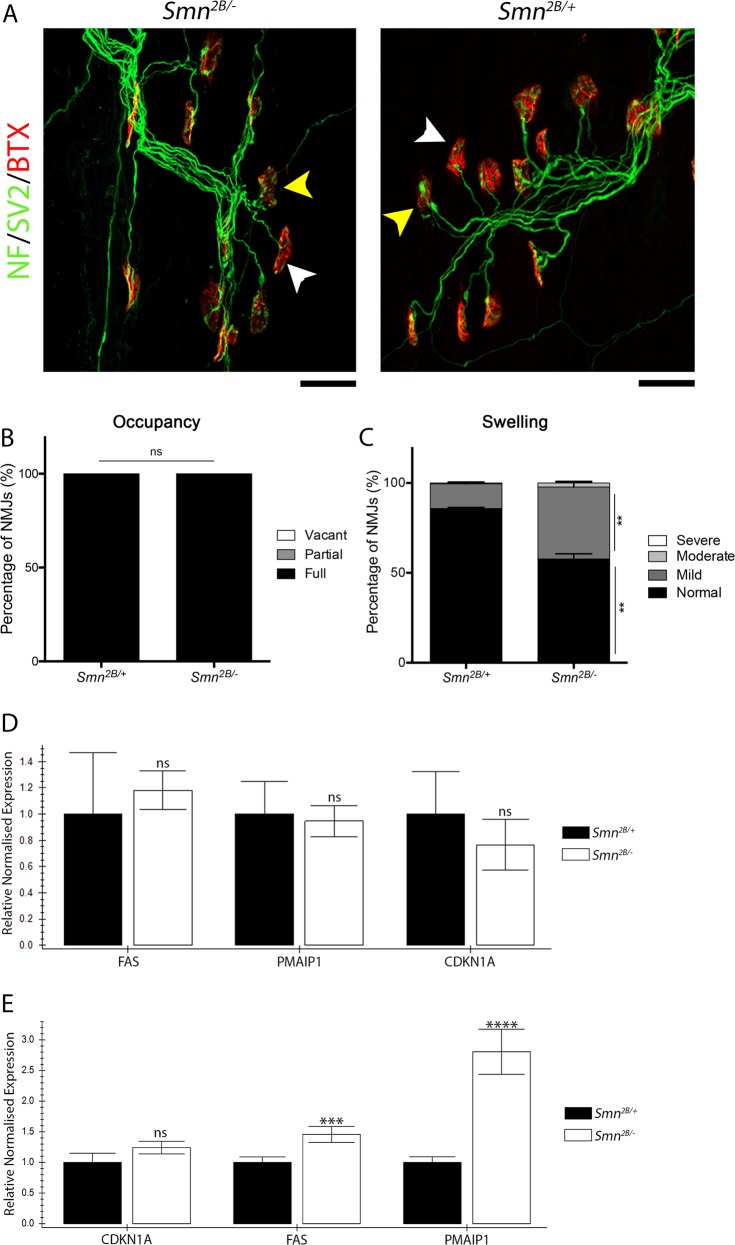
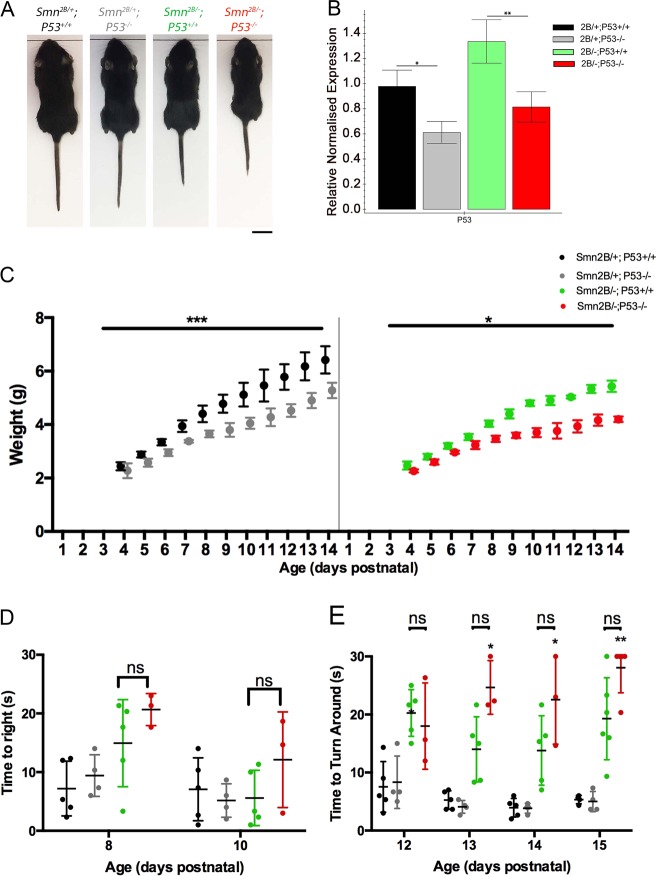
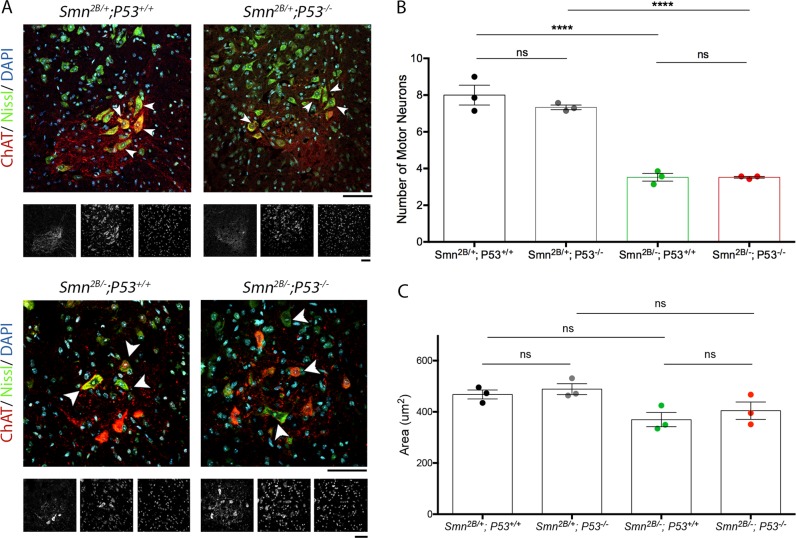
Spinal Muscular Atrophy (SMA) is a childhood motor neuron disease caused by mutations or deletions within the SMN1 gene. At endstages of disease there is profound loss of motor neurons, loss of axons within ventral roots and defects at the neuromuscular junctions (NMJ), as evidenced by pathological features such as pre-synaptic loss and swelling and post-synaptic shrinkage. Although these motor unit defects have been widely described, the time course and interdependancy of these aspects of motor unit degeneration are unclear. Recent reports have also revealed an early upregulation of transcripts associated with the P53 signalling pathway. The relationship between the upregulation of these transcripts and pathology within the motor unit is also unclear. In this study, we exploit the prolonged disease timecourse and defined pre-symptomatic period in the Smn mouse model to perform a temporal analysis of the different elements of motor unit pathology. We demonstrate that NMJ loss occurs prior to cell body loss, and coincides with the onset of symptoms. The onset of NMJ pathology also coincides with an increase in P53-related transcripts at the cell body. Finally, using a tamoxifen inducible P53 knockout, we demonstrate that post-natal reduction in P53 levels can reduce NMJ loss, but does not affect other aspects of NMJ pathology, motor neuron loss or the phenotype of the Smn mouse model. Together this work provides a detailed temporal description of pathology within motor units of an SMA mouse model, and demonstrates that NMJ loss is a P53-dependant process. This work supports the role for P53 as an effector of synaptic and axonal degeneration in a die-back neuropathy.
脊髓性肌萎缩症(SMA)是一种儿童运动神经元疾病,由 SMN1 基因的突变或缺失引起。在疾病的终末期,运动神经元严重丧失,腹根内的轴突丧失,神经肌肉接头(NMJ)出现缺陷,这可以通过前突触丧失和肿胀以及后突触收缩等病理特征来证明。尽管这些运动单位缺陷已被广泛描述,但这些运动单位退化方面的时间进程和相互依存性尚不清楚。最近的报告还揭示了与 P53 信号通路相关的转录本的早期上调。这些转录本的上调与运动单位内的病理学之间的关系也不清楚。在这项研究中,我们利用 Smn 小鼠模型中疾病的延长时间进程和明确的无症状前阶段,对运动单位病理学的不同方面进行了时间分析。我们证明 NMJ 的丧失发生在细胞体丧失之前,并且与症状发作同时发生。NMJ 病理学的发生也与细胞体中与 P53 相关的转录本增加同时发生。最后,使用他莫昔芬诱导的 P53 敲除,我们证明了出生后 P53 水平的降低可以减少 NMJ 的丧失,但不会影响 NMJ 病理学的其他方面、运动神经元的丧失或 Smn 小鼠模型的表型。总之,这项工作提供了 SMA 小鼠模型运动单位内病理学的详细时间描述,并证明了 NMJ 的丧失是一个依赖 P53 的过程。这项工作支持 P53 作为神经退行性疾病中突触和轴突退化的效应因子的作用。