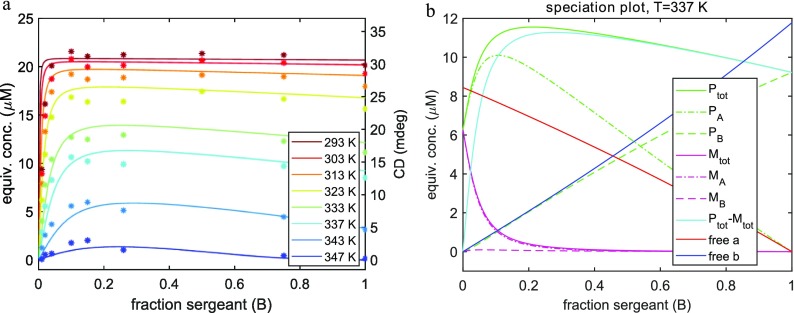
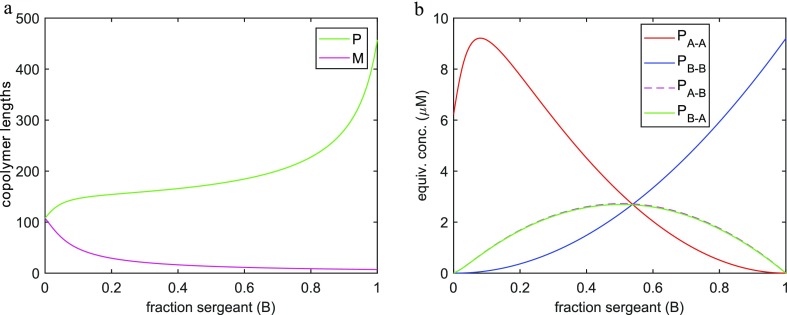
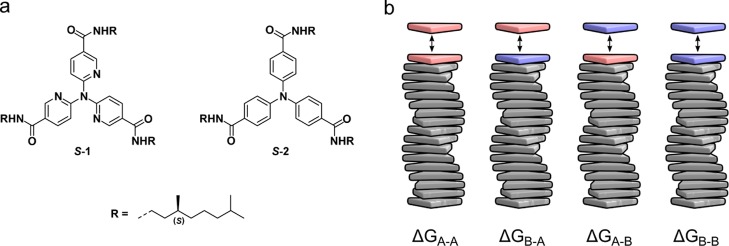
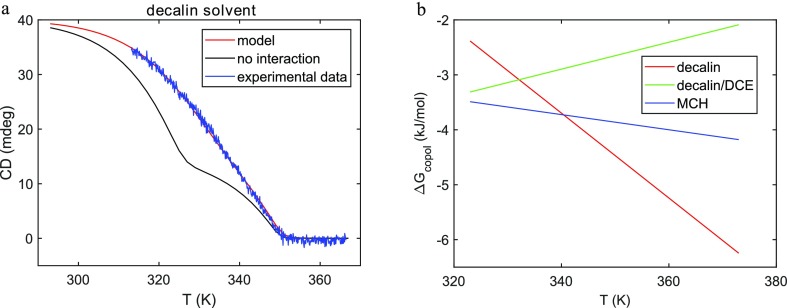
J Phys Chem B. 2019 Aug 1;123(30):6627-6642. doi: 10.1021/acs.jpcb.9b04373. Epub 2019 Jul 9.
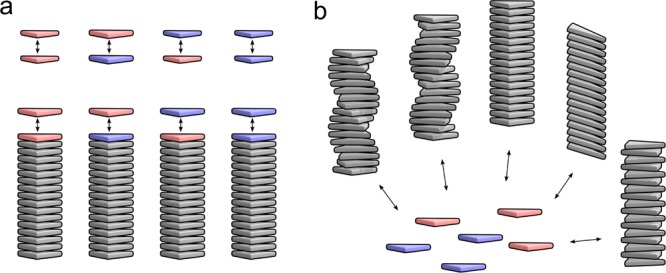
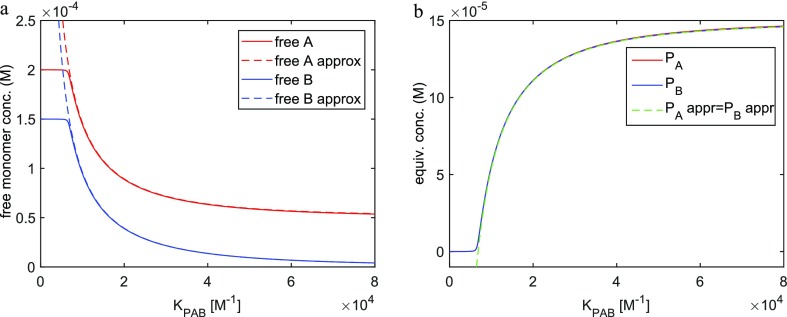
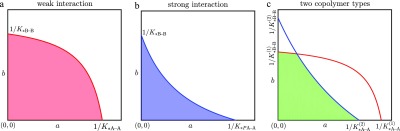
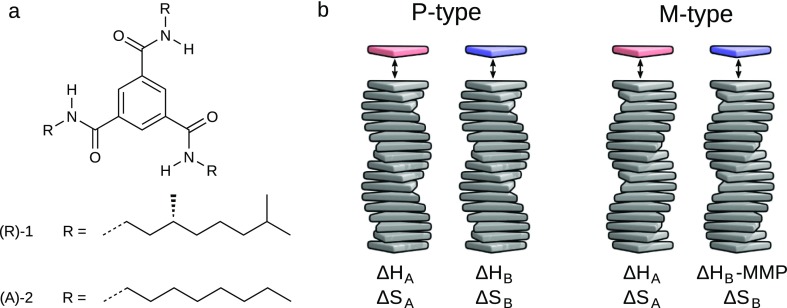
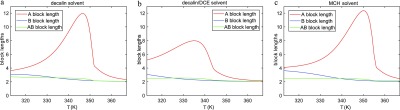
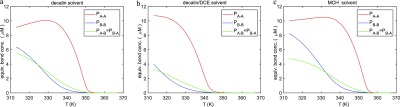
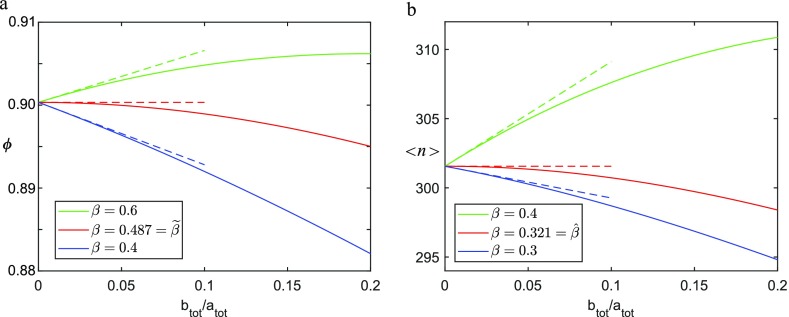
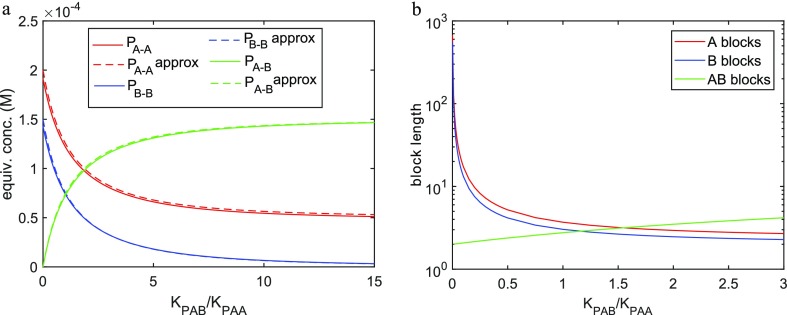
The coassembly of different building blocks into supramolecular copolymers provides a promising avenue to control their properties and to thereby expand the potential of supramolecular polymers in applications. However, contrary to covalent copolymerization which nowadays can be well controlled, the control over sequence, polymer length, and morphology in supramolecular copolymers is to date less developed, and their structures are more determined by the delicate balance in binding free energies between the distinct building blocks than by kinetics. Consequently, to rationalize the structures of supramolecular copolymers, a thorough understanding of their thermodynamic behavior is needed. Though this is well established for single-component assemblies and over the past years several models have been proposed for specific copolymerization cases, a generally applicable model for supramolecular cooperative copolymers is still lacking. Here, we provide a generalization of our earlier mass-balance models for supramolecular copolymerizations that encompasses all our earlier models. In this model, the binding free energies of each pair of monomer types in each aggregate type can be set independently. We provide scripts to solve the model numerically for any (co)polymerization of one or two types of monomer into an arbitrary number of distinct aggregate types. We illustrate the applicability of the model on data from literature as well as on new experimental data of triarylamine triamide-based copolymers in three distinct solvents. We show that apart from common properties such as the degree of polymerization and length distributions, our approach also allows us to investigate properties such as the copolymer microstructure, that is, the internal ordering of monomers within the copolymers. Moreover, we show that in some cases, also intriguing analytical approximations can be derived from the mass balances.
不同构建块的共组装形成超分子共聚物,为控制其性质并扩展超分子聚合物在应用中的潜力提供了有前景的途径。然而,与现今可以很好控制的共价共聚反应相反,对超分子共聚物中序列、聚合物长度和形态的控制至今还不够发达,其结构更多地取决于不同构建块之间结合自由能的微妙平衡,而不是动力学。因此,为了合理化超分子共聚物的结构,需要深入了解其热力学行为。尽管这对于单一组分组装已经得到很好的确立,并且在过去的几年中已经提出了几种针对特定共聚反应情况的模型,但仍然缺乏用于超分子协同共聚物的通用模型。在这里,我们提供了我们之前用于超分子共聚反应的质量平衡模型的推广,该模型涵盖了我们之前的所有模型。在这个模型中,可以独立设置每个聚合体型中每对单体类型的结合自由能。我们提供了用于数值求解模型的脚本,可用于任何(共)聚合反应,即将一种或两种类型的单体聚合到任意数量的不同聚合体型中。我们将模型应用于文献中的数据以及基于三芳基胺三酰胺的共聚物在三种不同溶剂中的新实验数据。我们表明,除了常见的性质,如聚合度和长度分布外,我们的方法还允许我们研究共聚物的微观结构等性质,即单体在共聚物中的内部排列。此外,我们表明,在某些情况下,还可以从质量平衡中推导出有趣的分析近似。