Department of Hepatopancreatobiliary Surgery, Cancer Hospital of China Medical University, Shenyang, Liaoning 110042, P.R. China.
Mol Med Rep. 2019 Sep;20(3):2519-2532. doi: 10.3892/mmr.2019.10487. Epub 2019 Jul 11.
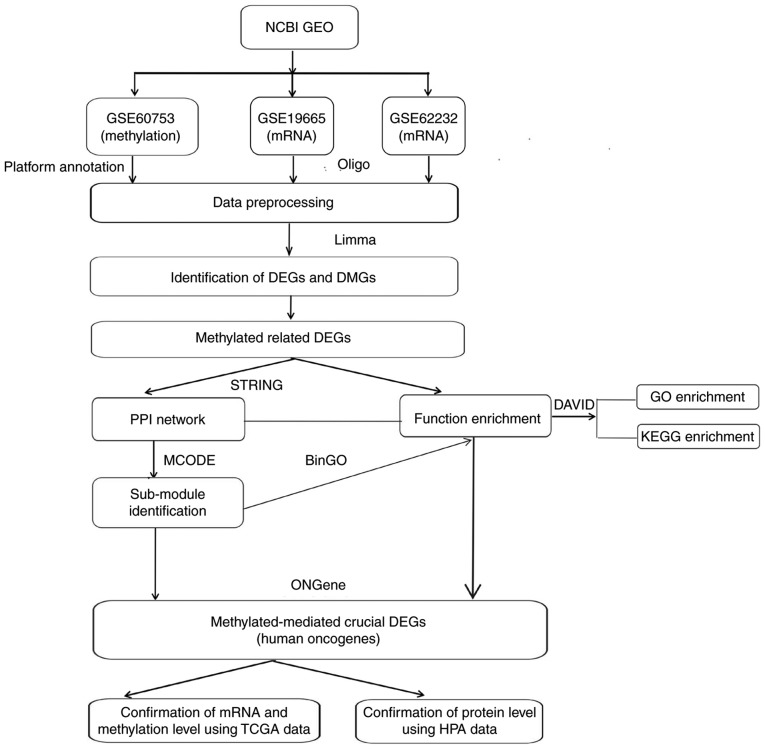
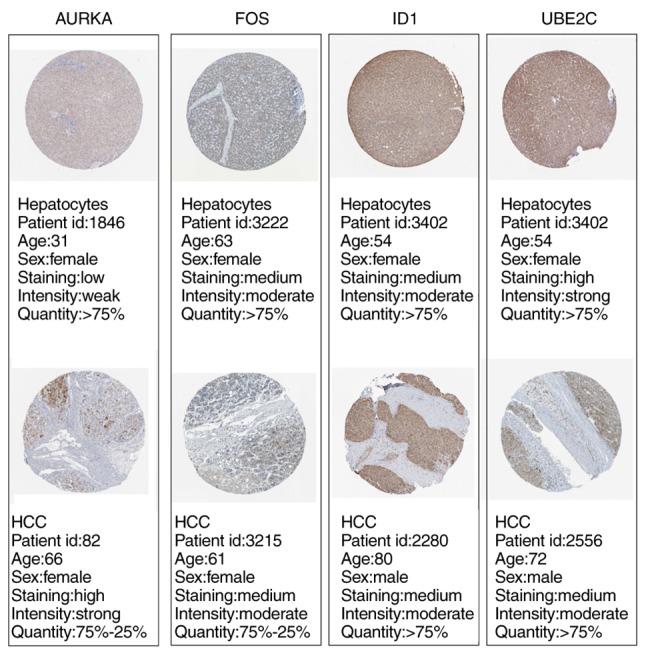
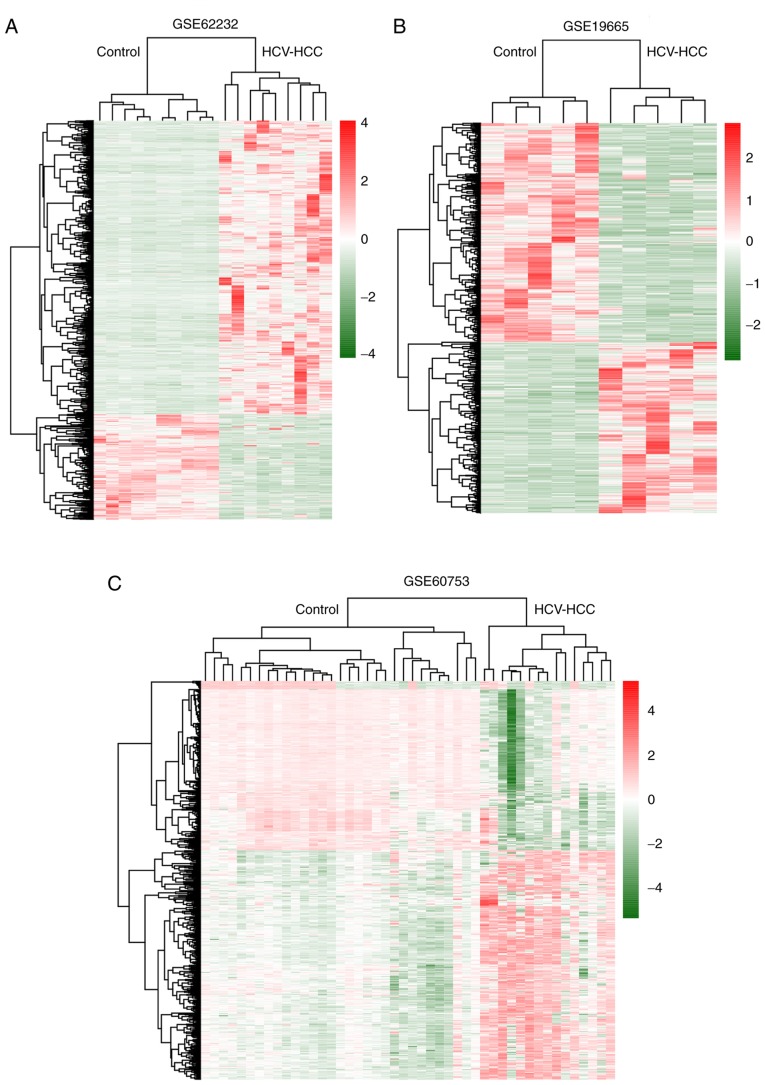
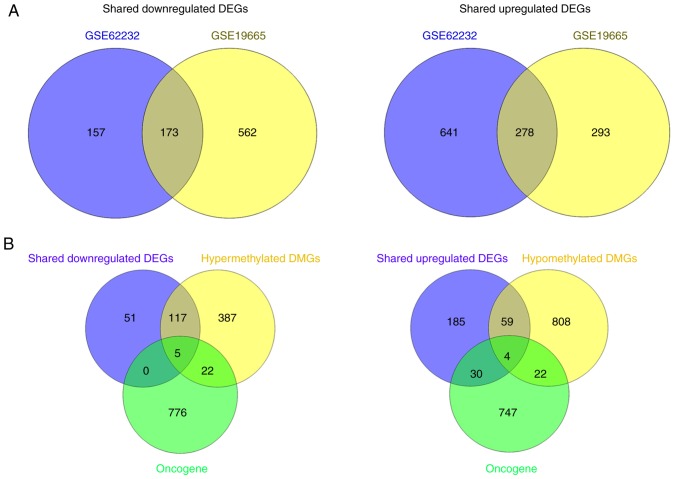
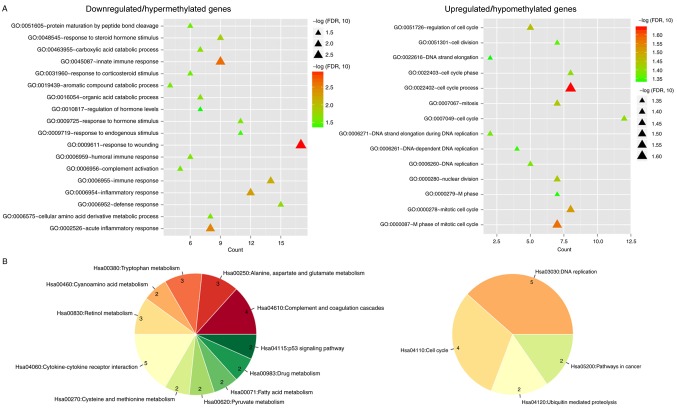
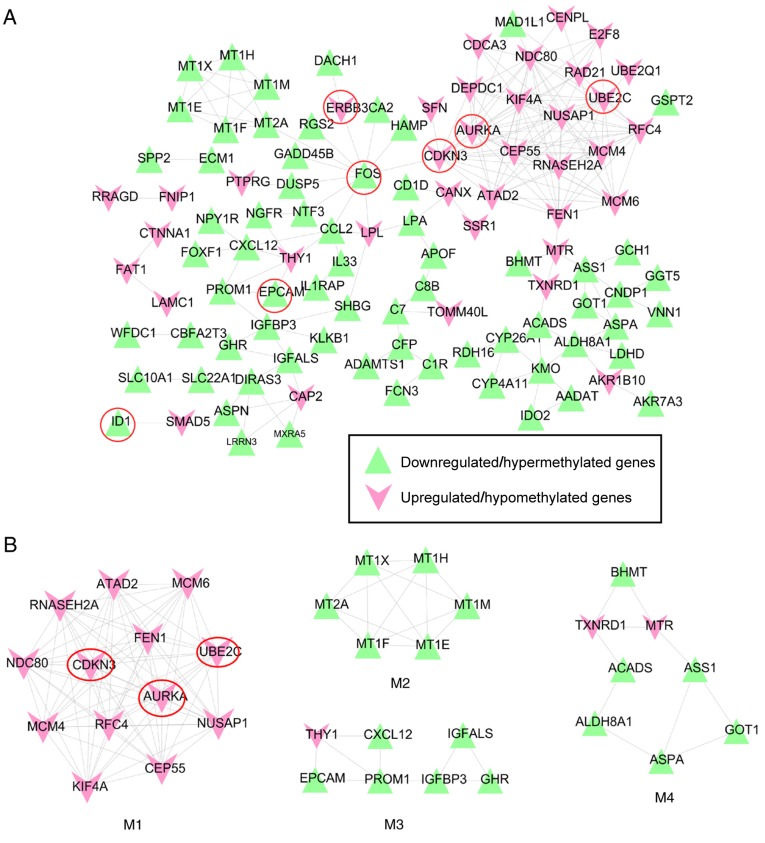
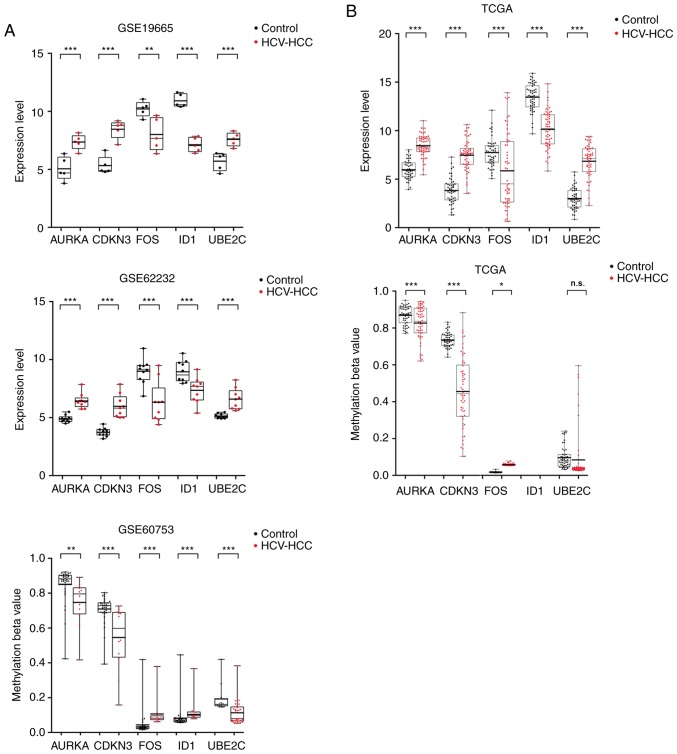
Changes in the methylation levels of tumor suppressor genes or proto‑oncogenes are involved in the pathogenesis of hepatitis C virus (HCV) infection‑induced hepatocellular carcinoma (HCC). The aim of the present study was to identify novel aberrantly methylated differentially expressed genes by integrating mRNA expression profile (GSE19665 and GSE62232) and methylation profile (GSE60753) microarrays downloaded from the Gene Expression Omnibus database. Functional enrichment analysis of screened genes was performed using the DAVID software and BinGO database. Protein‑protein interaction (PPI) networks were constructed using the STRING database, followed by module analysis with MCODE software. The transcriptional and translational expression levels of crucial genes were confirmed using The Cancer Genome Atlas (TCGA) datasets and Human Protein Atlas database (HPA). A total of 122 downregulated/hypermethylated genes and 63 upregulated/hypomethylated genes were identified. These genes were enriched in the Gene Ontology biological processes terms of 'inflammatory response' [Fos proto‑oncogene, AP‑1 transcription factor subunit (FOS)] and 'cell cycle process' [aurora kinase A (AURKA), cyclin dependent kinase inhibitor 3 (CDKN3) and ubiquitin conjugating enzyme E2 C (UBE2C)]. PPI network and module analysis indicated that human oncogenes FOS, AURKA, CDKN3 and UBE2C may be hub genes. mRNA, protein expression and methylation levels of AURKA and FOS were validated by TCGA and HPA data. In conclusion, aberrantly methylated AURKA and FOS may be potential therapeutic targets for HCV‑positive HCC.
抑癌基因或原癌基因的甲基化水平变化与丙型肝炎病毒(HCV)感染诱导的肝细胞癌(HCC)的发病机制有关。本研究旨在通过整合从基因表达综合数据库(GEO)下载的 mRNA 表达谱(GSE19665 和 GSE62232)和甲基化谱(GSE60753)微阵列,来鉴定新型异常甲基化的差异表达基因。使用 DAVID 软件和 BinGO 数据库对筛选出的基因进行功能富集分析。使用 STRING 数据库构建蛋白质-蛋白质相互作用(PPI)网络,然后使用 MCODE 软件进行模块分析。使用癌症基因组图谱(TCGA)数据集和人类蛋白质图谱数据库(HPA)验证关键基因的转录和翻译表达水平。共鉴定出 122 个下调/高甲基化基因和 63 个上调/低甲基化基因。这些基因富集于基因本体论生物过程术语“炎症反应”[原癌基因 Fos,AP-1 转录因子亚基(FOS)]和“细胞周期过程”[极光激酶 A(AURKA)、细胞周期蛋白依赖性激酶抑制剂 3(CDKN3)和泛素连接酶 E2 C(UBE2C)]。PPI 网络和模块分析表明,人类癌基因 FOS、AURKA、CDKN3 和 UBE2C 可能是枢纽基因。通过 TCGA 和 HPA 数据验证了 AURKA 和 FOS 的 mRNA、蛋白表达和甲基化水平。总之,异常甲基化的 AURKA 和 FOS 可能是 HCV 阳性 HCC 的潜在治疗靶点。