Department of Infectious Diseases, Baoji Municipal Central Hospital, Baoji, Shaanxi 721008, P.R. China.
Mol Med Rep. 2017 Nov;16(5):6674-6682. doi: 10.3892/mmr.2017.7457. Epub 2017 Sep 11.
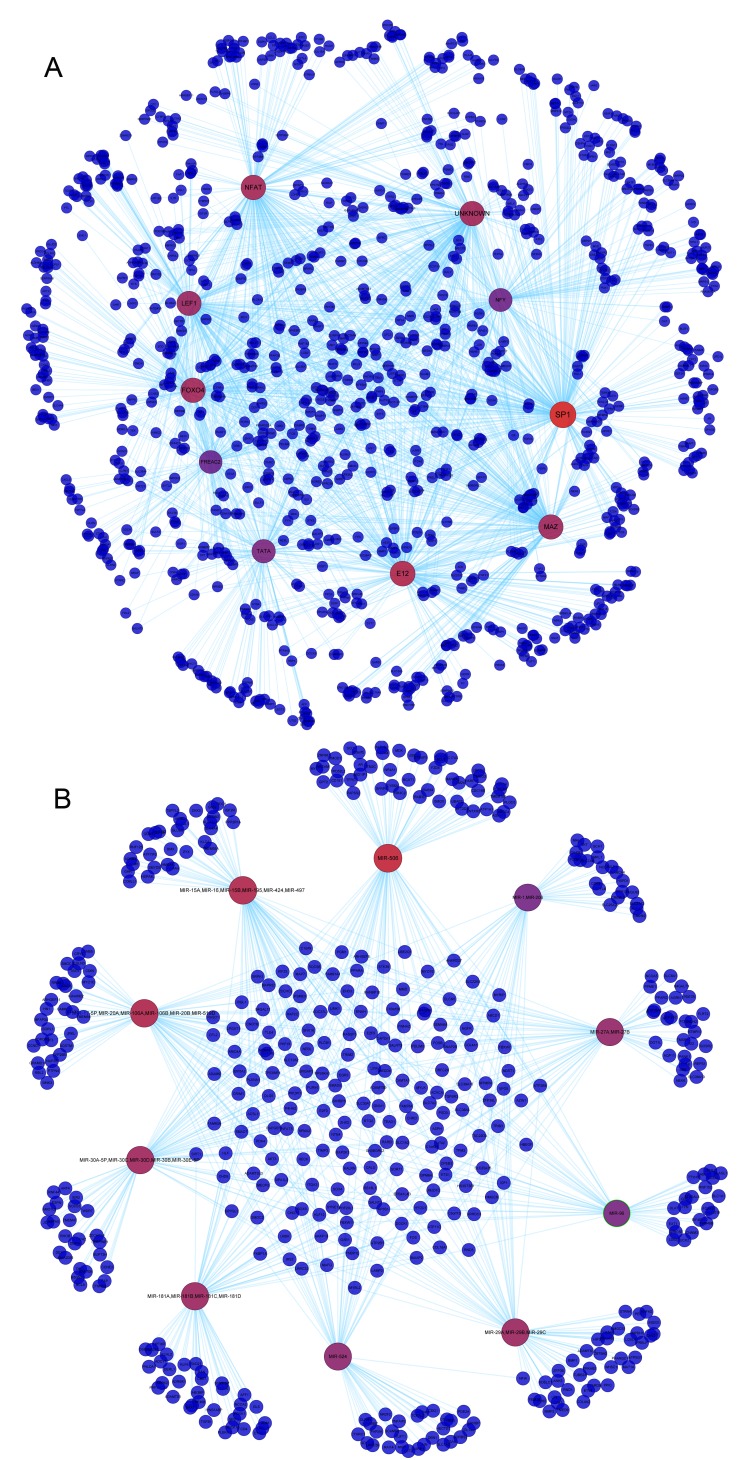
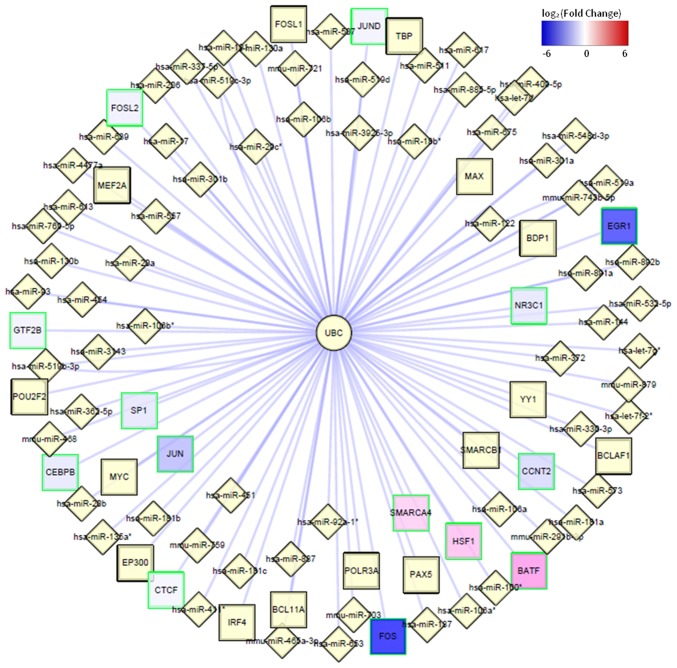
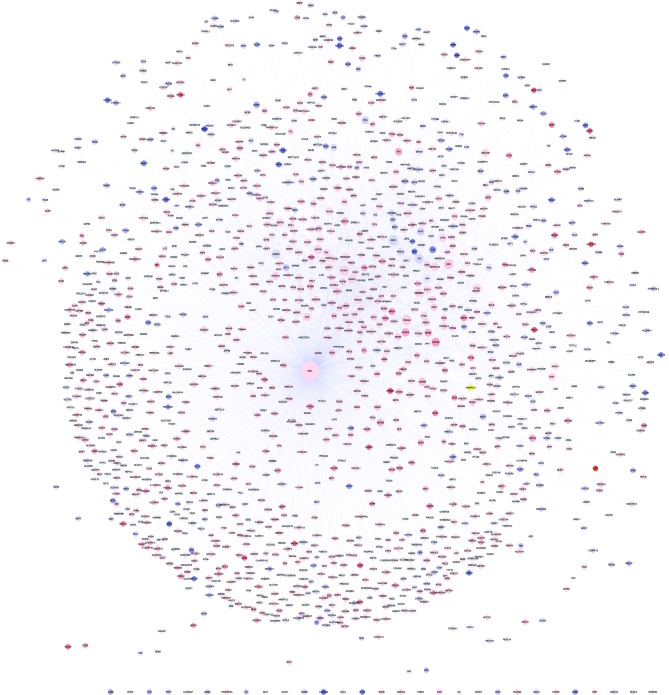
The present study aimed to explore the underlying molecular mechanisms of hepatocellular carcinoma (HCC). RNA‑sequencing profiles GSM629264 and GSM629265, from the GSE25599 data set, were downloaded from the Gene Expression Omnibus database and processed by quality evaluation. GSM629264 and GSM629265 were from HCC and adjacent non‑cancerous tissues, respectively. TopHat software was used for alignment analysis, followed by the detection of novel splicing sites. In addition, the Cufflinks software package was used to analyze gene expressions, and the Cuffdiff program was used to screen for differently expressed genes (DEGs) and differentially expressed splicing variants. Gene ontology functional enrichment and Kyoto Encyclopedia of Genes and Genomes pathway enrichment analyses of DEGs were also performed. Transcription factors (TFs) and microRNAs (miRNAs) that regulate DEGs were identified, and a protein‑protein interaction (PPI) network was constructed. The hub node in the PPI network was obtained, and the TFs and miRNAs that regulated the hub node were further predicted. The quality of the sequencing data met the standards for analysis, and the clean reads were ~65%. Most sequencing reads mapped into coding sequence exons (CDS_exons), whereas other reads mapped into exon 3' untranslated regions (UTR_Exons), 5'UTR_Exons and Introns. Upregulated and downregulated DEGs between HCC and adjacent non‑cancerous tissues were screened. Genes of differentially expressed splicing variants were identified, including vesicle‑associated membrane protein 4, phosphatidylinositol glycan anchor biosynthesis class C, protein disulfide isomerase family A member 4 and growth arrest specific 5. Screened DEGs were enriched in the complement pathway. In the PPI network, ubiquitin C (UBC) was the hub node. UBC was predicted to be regulated by several TFs, including specificity protein 1 (SP1), FBJ murine osteosarcoma viral oncogene homolog (FOS), proto‑oncogene c‑JUN (JUN), FOS‑like antigen 2 (FOSL2) and SWI/SNF‑related, matrix‑associated, actin‑dependent regulator of chromatin, subfamily A, member 4 (SMARCA4), and several miRNAs, including miR‑30 and miR‑181. Results from the present study demonstrated that UBC, SP1, FOS, JUN, FOSL2, SMARCA4, miR‑30 and miR‑181 may participate in the development of HCC.
本研究旨在探索肝细胞癌(HCC)的潜在分子机制。从 Gene Expression Omnibus 数据库中下载 GSE25599 数据集的 RNA-seq 谱 GSM629264 和 GSM629265,经过质量评估后进行处理。GSM629264 和 GSM629265 分别来自 HCC 和相邻的非癌组织。使用 TopHat 软件进行比对分析,随后检测新的剪接位点。此外,使用 Cufflinks 软件包分析基因表达,并使用 Cuffdiff 程序筛选差异表达基因(DEGs)和差异表达剪接变体。还对 DEGs 进行了基因本体功能富集和京都基因与基因组百科全书通路富集分析。鉴定调控 DEGs 的转录因子(TFs)和 microRNAs(miRNAs),构建蛋白质-蛋白质相互作用(PPI)网络。获得 PPI 网络中的枢纽节点,并进一步预测调控枢纽节点的 TFs 和 miRNAs。测序数据的质量符合分析标准,清洁读数约为 65%。大多数测序读数映射到编码序列外显子(CDS_exons),而其他读数映射到外显子 3'非翻译区(UTR_Exons)、5'UTR_Exons 和内含子。筛选 HCC 和相邻非癌组织之间的上调和下调 DEGs。鉴定差异表达剪接变体的基因,包括囊泡相关膜蛋白 4、磷酸肌醇聚糖锚生物合成类 C、蛋白二硫键异构酶家族 A 成员 4 和生长停滞特异性 5。筛选的 DEGs 富集在补体途径中。在 PPI 网络中,泛素 C(UBC)是枢纽节点。预测 UBC 受几个 TFs 调控,包括特异性蛋白 1(SP1)、FBJ 鼠骨肉瘤病毒癌基因同源物(FOS)、原癌基因 c-JUN(JUN)、FOS 样抗原 2(FOSL2)和 SWI/SNF 相关、基质相关、肌动蛋白依赖性染色质调节剂亚家族 A,成员 4(SMARCA4)和几个 miRNAs,包括 miR-30 和 miR-181。本研究结果表明,UBC、SP1、FOS、JUN、FOSL2、SMARCA4、miR-30 和 miR-181 可能参与 HCC 的发生发展。