Department of Physiology & Pathophysiology, Fujian Medical University, Fuzhou, China.
The Key Laboratory of Fujian Province University on Ion Channel and Signal Transduction in Cardiovascular Disease, School of Basic Medical Sciences, Fujian Medical University, Fuzhou, China.
J Transl Med. 2019 Jul 22;17(1):231. doi: 10.1186/s12967-019-1981-5.
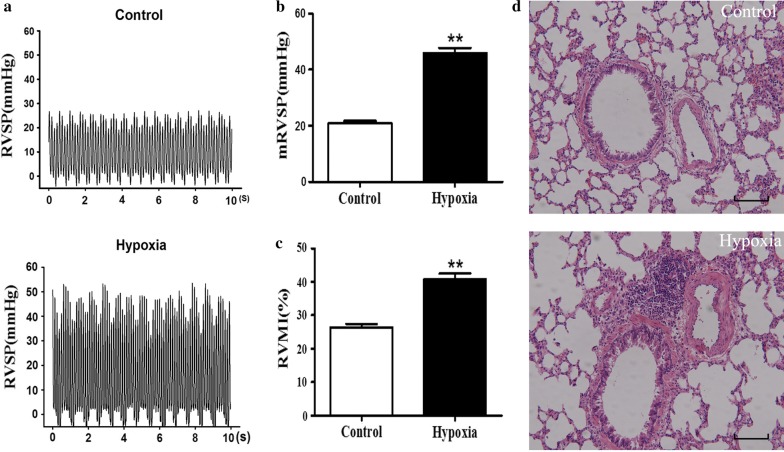
Pulmonary artery hypertension (PAH), which is characterized by an increase in pulmonary circulation blood pressure, is a fatal disease, and its pathogenesis remains unclear.
In this study, RNA sequencing (RNA-seq), tandem mass tags (TMT) and reduced representation bisulfite sequencing (RRBS) were performed to detect the levels of mRNA, protein, and DNA methylation in pulmonary arteries (PAs), respectively. To screen the possible pathways and proteins related to PAH, pathway enrichment analysis and protein-protein interaction (PPI) network analysis were performed. For selected genes, differential expression levels were confirmed at both the transcriptional and translational levels by real-time PCR and Western blot analyses, respectively.
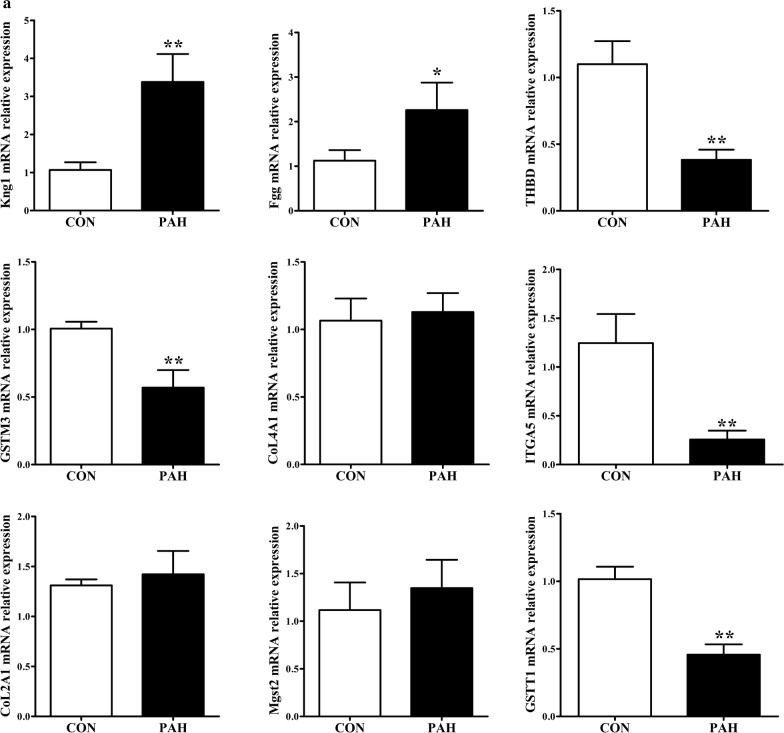
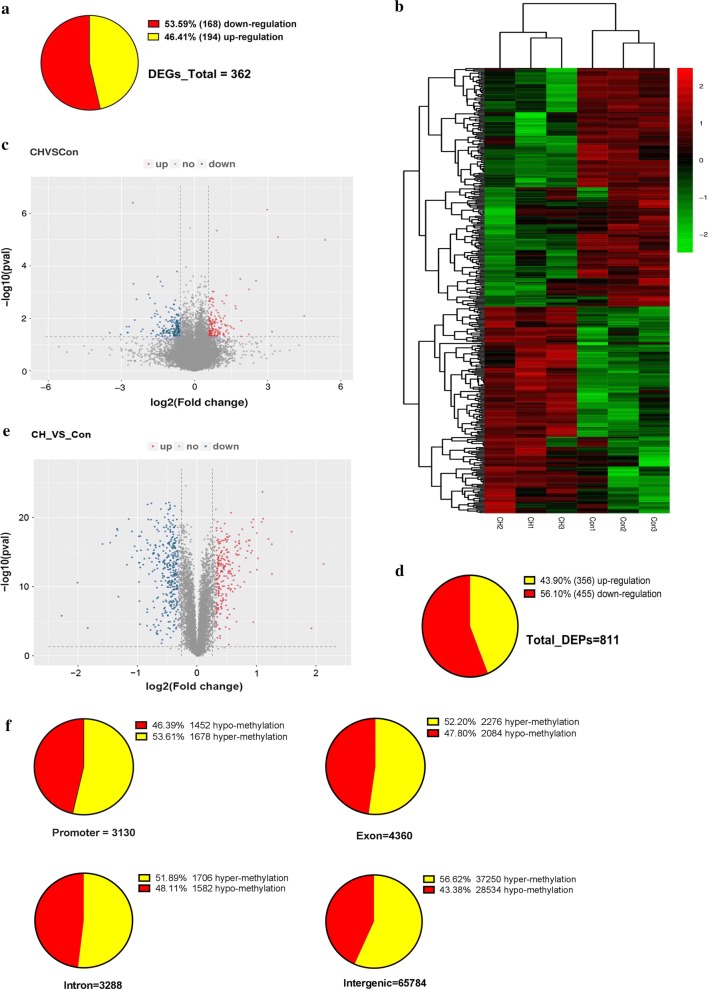
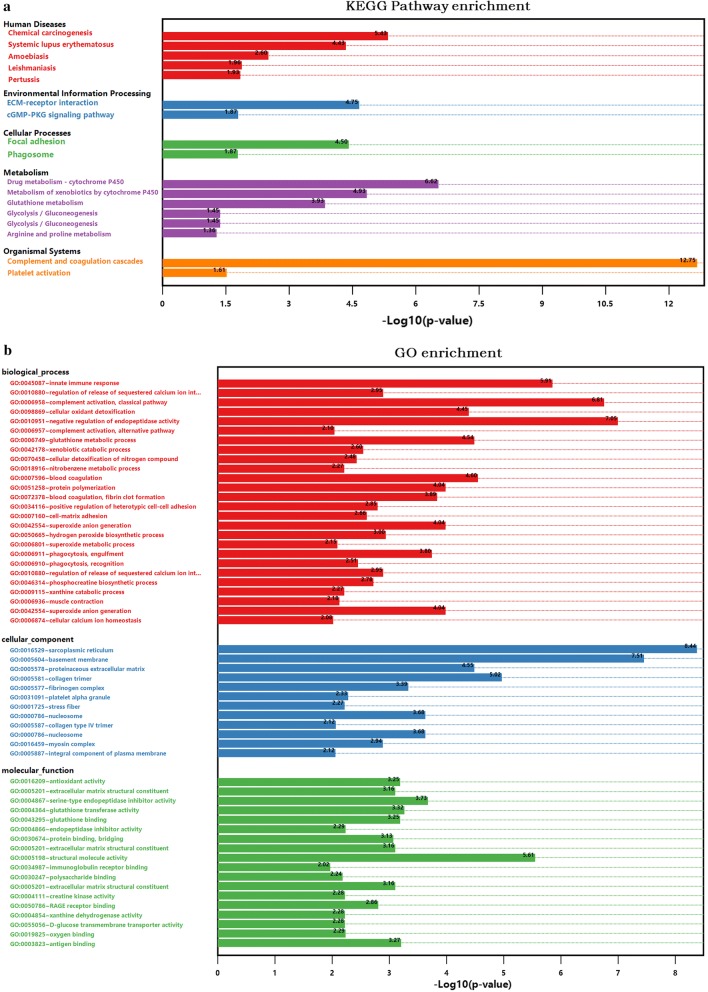
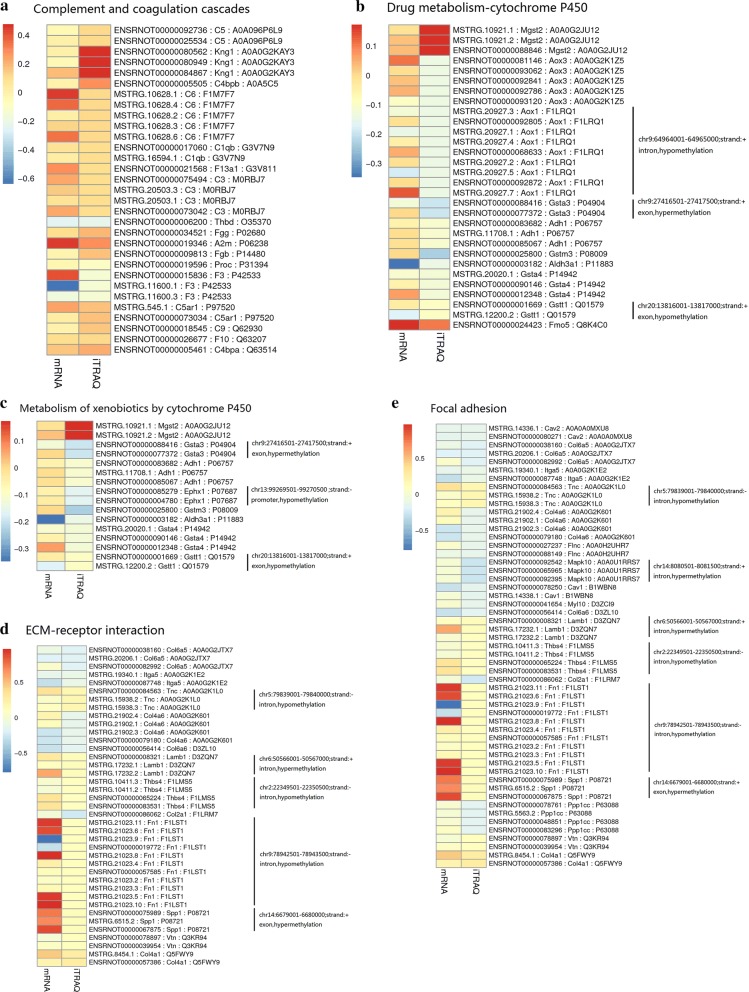
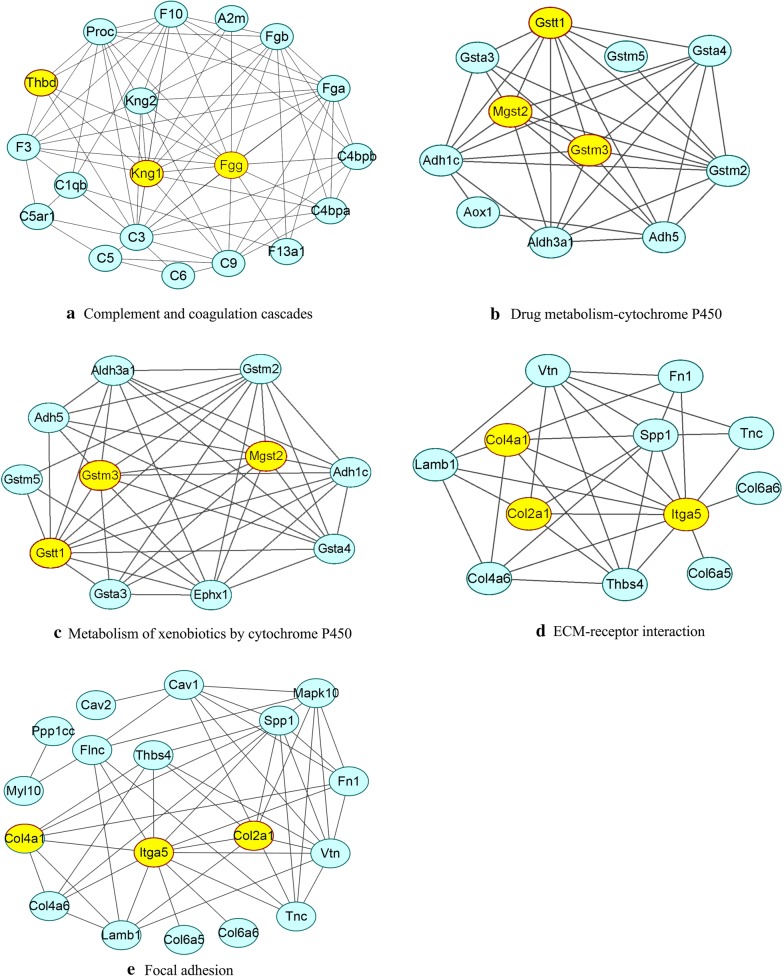
A total of 362 differentially expressed genes (|Fold-change| > 1.5 and p < 0.05), 811 differentially expressed proteins (|Fold-change| > 1.2 and p < 0.05) and 76,562 differentially methylated regions (1000 bp slide windows, 500 bp overlap, p < 0.05, and |Fold-change| > 1.2) were identified when the PAH group (n = 15) was compared with the control group (n = 15). Through an integrated analysis of the characteristics of the three omic analyses, a multiomics table was constructed. Additionally, pathway enrichment analysis showed that the differentially expressed proteins were significantly enriched in five Kyoto Encyclopedia of Genes and Genomes (KEGG) biological pathways and ten Gene Ontology (GO) terms for the PAH group compared with the control group. Moreover, protein-protein interaction (PPI) networks were constructed to identify hub genes. Finally, according to the genes identified in the PPI and the protein expression fold-change, nine key genes and their associated proteins were verified by real-time PCR and Western blot analyses, including Col4a1, Itga5, Col2a1, Gstt1, Gstm3, Thbd, Mgst2, Kng1 and Fgg.
This study conducted multiomic characteristic profiling to identify genes that contribute to the hypoxia-induced PAH model, identifying new avenues for basic PAH research.
肺动脉高压(PAH)的特征是肺循环血压升高,是一种致命的疾病,其发病机制尚不清楚。
本研究分别采用 RNA 测序(RNA-seq)、串联质量标签(TMT)和简化重亚硫酸盐测序(RRBS)检测肺动脉(PA)中的 mRNA、蛋白质和 DNA 甲基化水平。为了筛选可能与 PAH 相关的途径和蛋白质,进行了途径富集分析和蛋白质-蛋白质相互作用(PPI)网络分析。对于选定的基因,通过实时 PCR 和 Western blot 分析分别在转录和翻译水平上确认差异表达水平。
当 PAH 组(n=15)与对照组(n=15)比较时,共鉴定出 362 个差异表达基因(|Fold-change|>1.5,p<0.05)、811 个差异表达蛋白(|Fold-change|>1.2,p<0.05)和 76562 个差异甲基化区域(1000 bp 滑动窗口,500 bp 重叠,p<0.05,|Fold-change|>1.2)。通过对三种组学特征的综合分析,构建了一个多组学表。此外,通路富集分析显示,与对照组相比,PAH 组差异表达蛋白在五个京都基因与基因组百科全书(KEGG)生物通路和十个基因本体论(GO)术语中显著富集。此外,构建蛋白质-蛋白质相互作用(PPI)网络以识别枢纽基因。最后,根据 PPI 中鉴定的基因和蛋白质表达倍数变化,通过实时 PCR 和 Western blot 分析验证了 9 个关键基因及其相关蛋白,包括 Col4a1、Itga5、Col2a1、Gstt1、Gstm3、Thbd、Mgst2、Kng1 和 Fgg。
本研究进行了多组学特征分析,以鉴定参与缺氧诱导的 PAH 模型的基因,为基础 PAH 研究开辟了新途径。