Centre for Gene Regulation and Expression, School of Life Sciences, University of Dundee, Dow St, Dundee, DD1 5EH, United Kingdom.
Centre for Gene Regulation and Expression, School of Life Sciences, University of Dundee, Dow St, Dundee, DD1 5EH, United Kingdom.
Mol Cell Proteomics. 2019 Oct;18(10):1967-1980. doi: 10.1074/mcp.RA119.001472. Epub 2019 Jul 22.
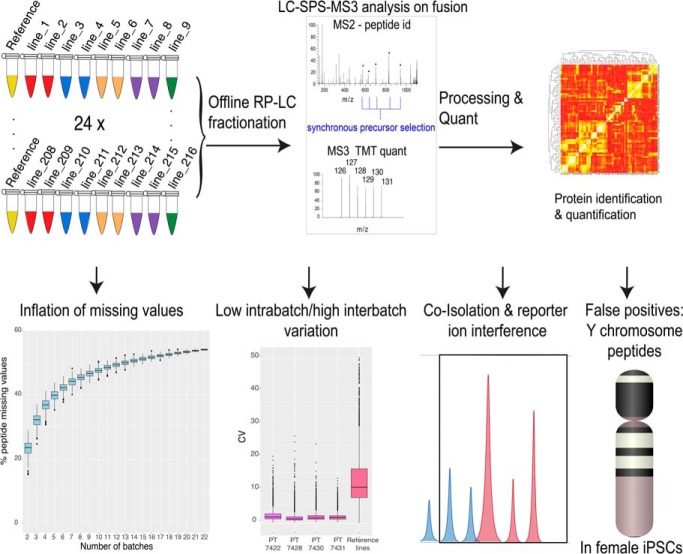
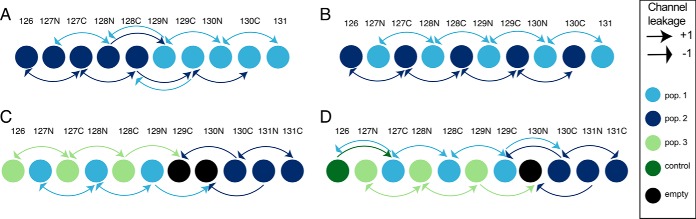
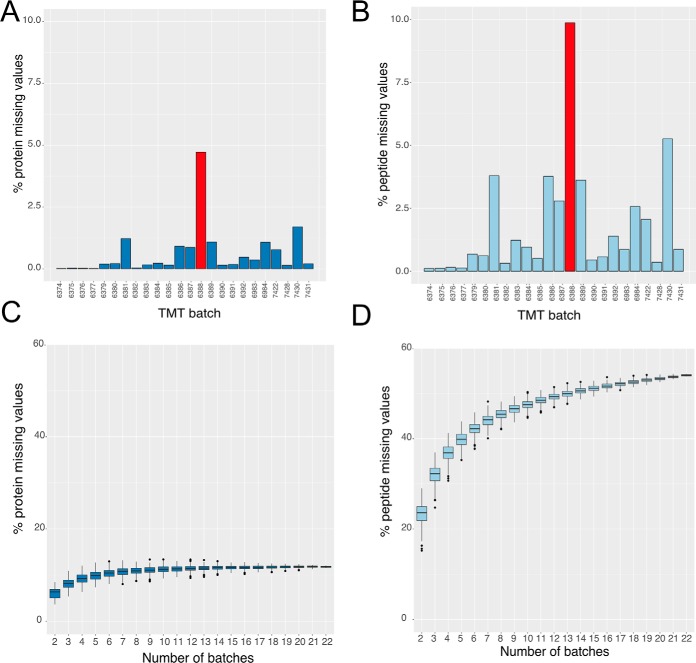
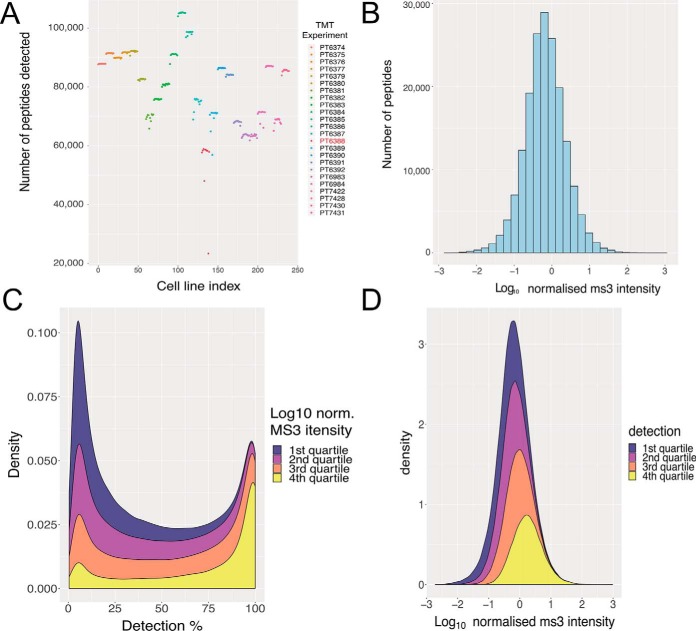
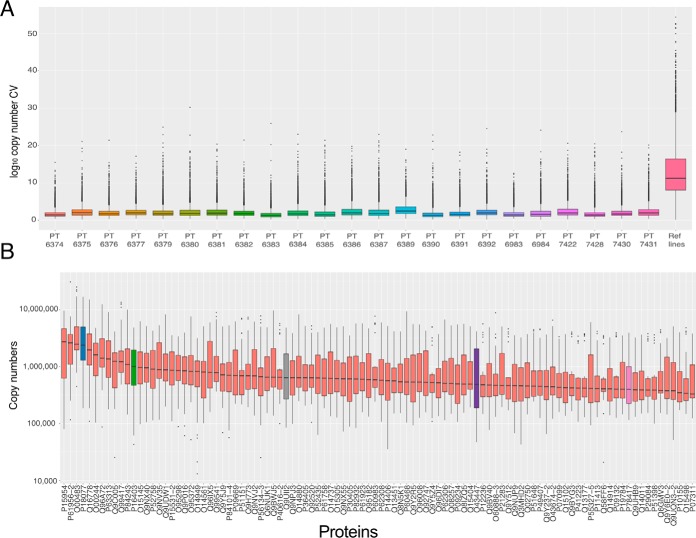
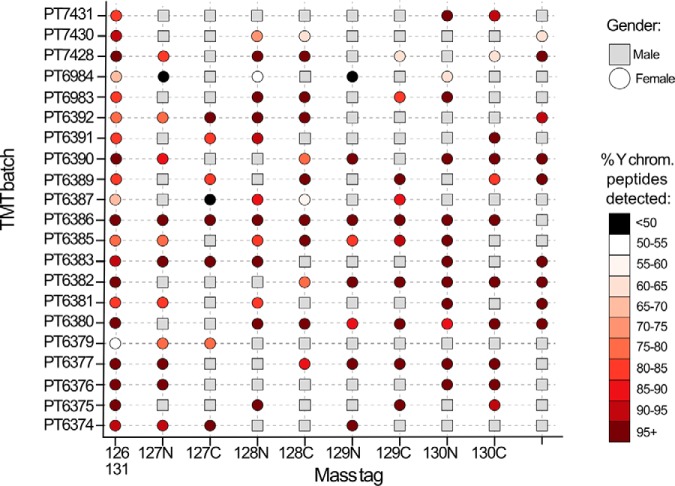
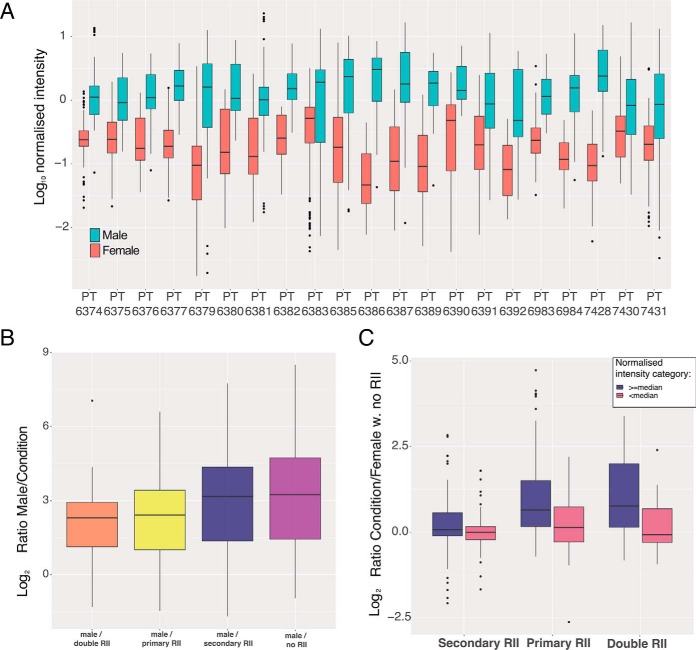
Multiplexing strategies for large-scale proteomic analyses have become increasingly prevalent, tandem mass tags (TMT) in particular. Here we used a large iPSC proteomic experiment with twenty-four 10-plex TMT batches to evaluate the effect of integrating multiple TMT batches within a single analysis. We identified a significant inflation rate of protein missing values as multiple batches are integrated and show that this pattern is aggravated at the peptide level. We also show that without normalization strategies to address the batch effects, the high precision of quantitation within a single multiplexed TMT batch is not reproduced when data from multiple TMT batches are integrated.Further, the incidence of false positives was studied by using Y chromosome peptides as an internal control. The iPSC lines quantified in this data set were derived from both male and female donors, hence the peptides mapped to the Y chromosome should be absent from female lines. Nonetheless, these Y chromosome-specific peptides were consistently detected in the female channels of all TMT batches. We then used the same Y chromosome specific peptides to quantify the level of ion coisolation as well as the effect of primary and secondary reporter ion interference. These results were used to propose solutions to mitigate the limitations of multi-batch TMT analyses. We confirm that including a common reference line in every batch increases precision by facilitating normalization across the batches and we propose experimental designs that minimize the effect of cross population reporter ion interference.
多重化策略在大规模蛋白质组学分析中越来越流行,串联质量标签(TMT)尤其如此。在这里,我们使用了一个包含二十四批十重 TMT 批处理的大型 iPSC 蛋白质组实验,来评估在单个分析中整合多个 TMT 批处理的效果。我们发现,随着多个批次的整合,蛋白质缺失值的发生率显著增加,并且这种模式在肽水平上更为严重。我们还表明,如果没有归一化策略来解决批次效应,那么当整合多个 TMT 批处理的数据时,单个多重 TMT 批处理内的高精度定量就无法重现。此外,我们还使用 Y 染色体肽作为内部对照来研究假阳性的发生率。该数据集定量的 iPSC 系来自男性和女性供体,因此映射到 Y 染色体的肽段应该不存在于女性系中。然而,在所有 TMT 批处理的女性通道中,这些 Y 染色体特异性肽段都被一致地检测到。然后,我们使用相同的 Y 染色体特异性肽段来定量离子共洗脱的水平,以及初级和次级报告离子干扰的影响。这些结果被用来提出解决方案,以减轻多批 TMT 分析的局限性。我们证实,在每个批次中包含一个通用参考线可以通过促进批次间的归一化来提高精度,并且我们提出了实验设计,以最小化跨群体报告离子干扰的影响。