Department of Medicine, University of Alberta, Edmonton, AB, T6G 2E1, Canada.
Department of Biological Sciences, University of Alberta, Edmonton, AB, T6G 2E9, Canada.
BMC Genomics. 2019 Jul 23;20(1):604. doi: 10.1186/s12864-019-5965-x.
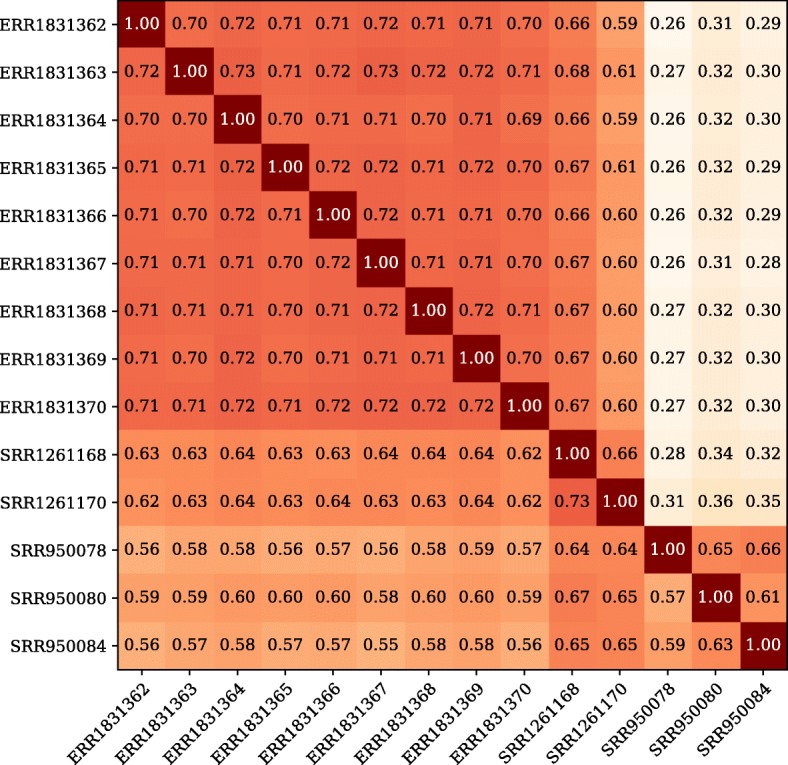
RNA-Seq data is inherently nonuniform for different transcripts because of differences in gene expression. This makes it challenging to decide how much data should be generated from each sample. How much should one spend to recover the less expressed transcripts? The sequencing technology used is another consideration, as there are inevitably always biases against certain sequences. To investigate these effects, we first looked at high-depth libraries from a set of well-annotated organisms to ascertain the impact of sequencing depth on de novo assembly. We then looked at libraries sequenced from the Universal Human Reference RNA (UHRR) to compare the performance of Illumina HiSeq and MGI DNBseq™ technologies.
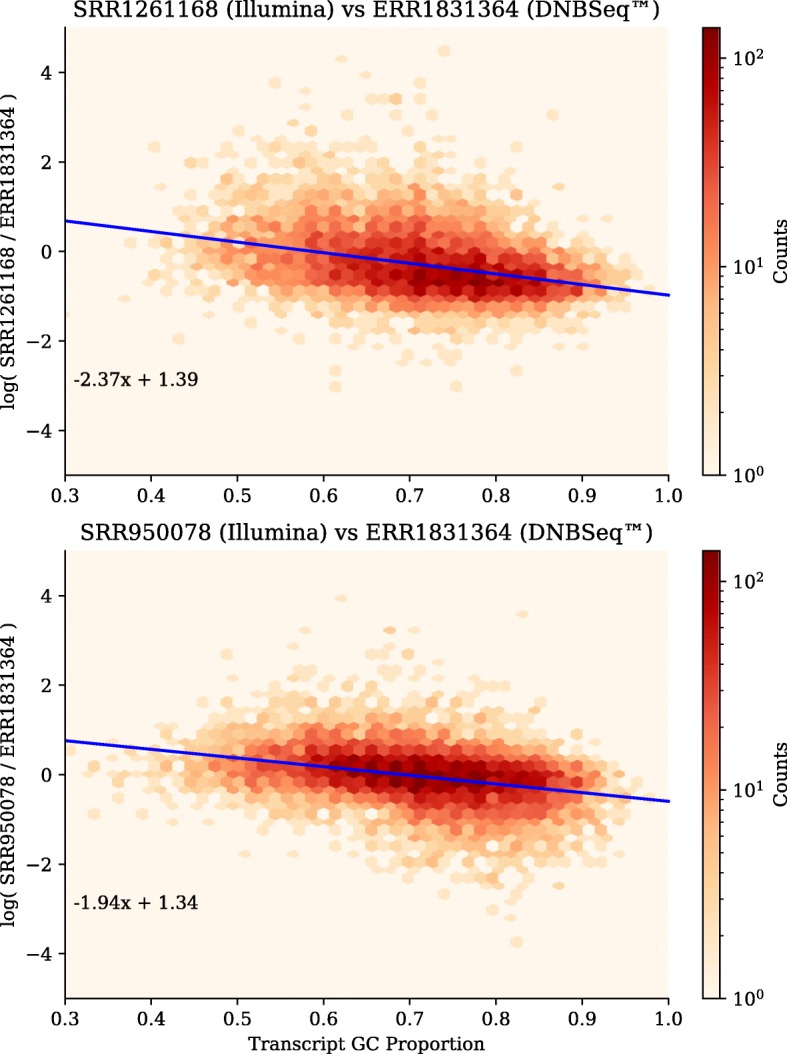
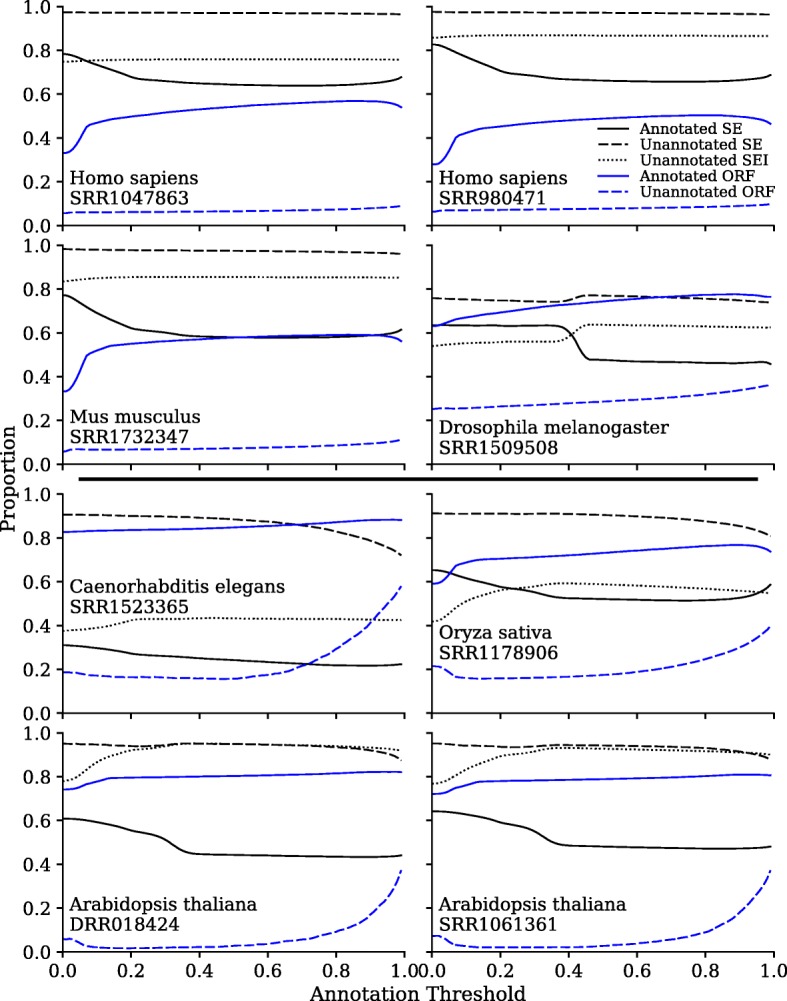
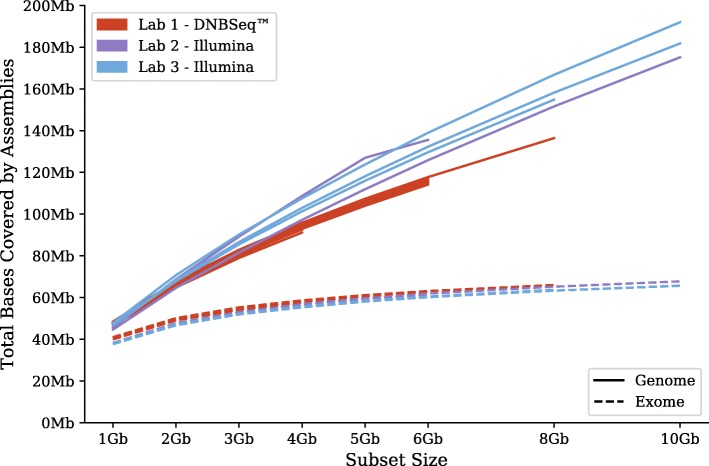
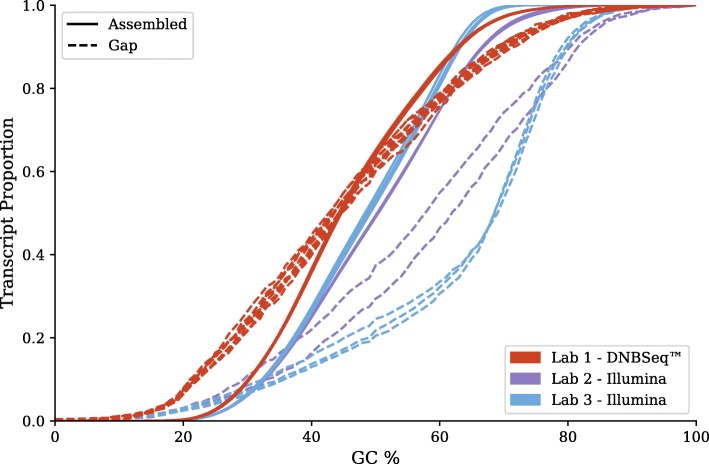
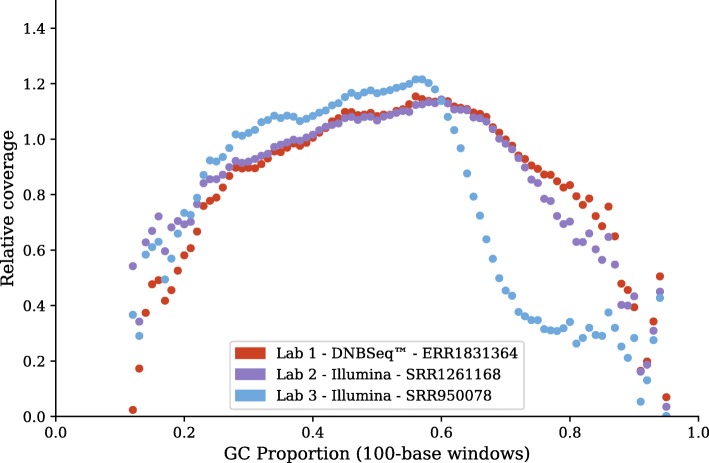
On the issue of sequencing depth, the amount of exomic sequence assembled plateaued using data sets of approximately 2 to 8 Gbp. However, the amount of genomic sequence assembled did not plateau for many of the analyzed organisms. Most of the unannotated genomic sequences are single-exon transcripts whose biological significance will be questionable for some users. On the issue of sequencing technology, both of the analyzed platforms recovered a similar number of full-length transcripts. The missing "gap" regions in the HiSeq assemblies were often attributed to higher GC contents, but this may be an artefact of library preparation and not of sequencing technology.
Increasing sequencing depth beyond modest data sets of less than 10 Gbp recovers a plethora of single-exon transcripts undocumented in genome annotations. DNBseq™ is a viable alternative to HiSeq for de novo RNA-Seq assembly.
由于基因表达的差异,不同转录本的 RNA-Seq 数据本质上是不均匀的。这使得很难确定应该从每个样本中生成多少数据。对于表达较少的转录本,应该花费多少来恢复它们?所使用的测序技术也是另一个需要考虑的因素,因为某些序列总是不可避免地存在偏见。为了研究这些影响,我们首先查看了一组经过良好注释的生物体的高深度文库,以确定测序深度对从头组装的影响。然后,我们查看了从通用人类参考 RNA(UHRR)测序的文库,以比较 Illumina HiSeq 和 MGI DNBseq™ 技术的性能。
关于测序深度的问题,使用大约 2 到 8 Gbp 的数据集组装的外显子序列量达到了平台期。然而,对于许多分析的生物体来说,组装的基因组序列量并没有达到平台期。大多数未注释的基因组序列是单外显子转录本,对于一些用户来说,它们的生物学意义将是值得怀疑的。关于测序技术的问题,两种分析平台都恢复了相似数量的全长转录本。HiSeq 组装中缺失的“缺口”区域通常归因于较高的 GC 含量,但这可能是文库制备而不是测序技术的人为产物。
在 10 Gbp 以下的适度数据集之外增加测序深度,会恢复大量在基因组注释中未记录的单外显子转录本。DNBseq™ 是从头 RNA-Seq 组装的 HiSeq 替代方案。