National Translational Science Center for Molecular Medicine, Department of Cell Biology, School of Basic Medicine, Air Force Medical University, Xi'an, China.
Cell Biology Program, Memorial Sloan-Kettering Cancer Center, New York, NY, USA.
Nature. 2019 Aug;572(7769):402-406. doi: 10.1038/s41586-019-1426-6. Epub 2019 Jul 24.
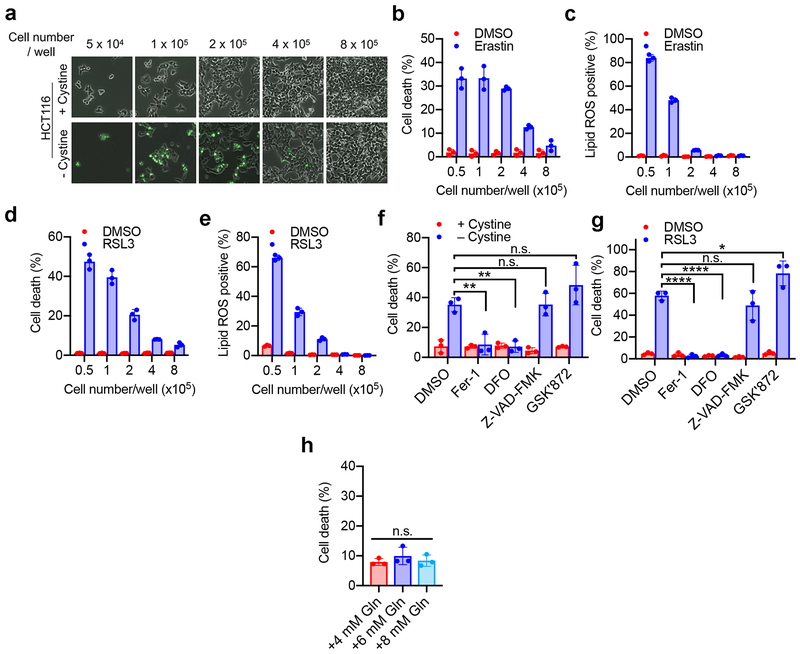
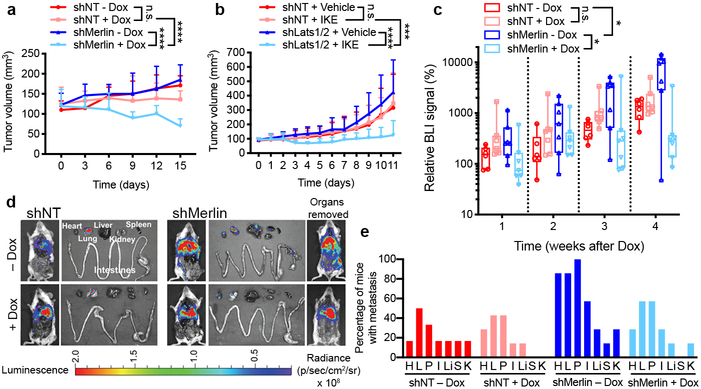
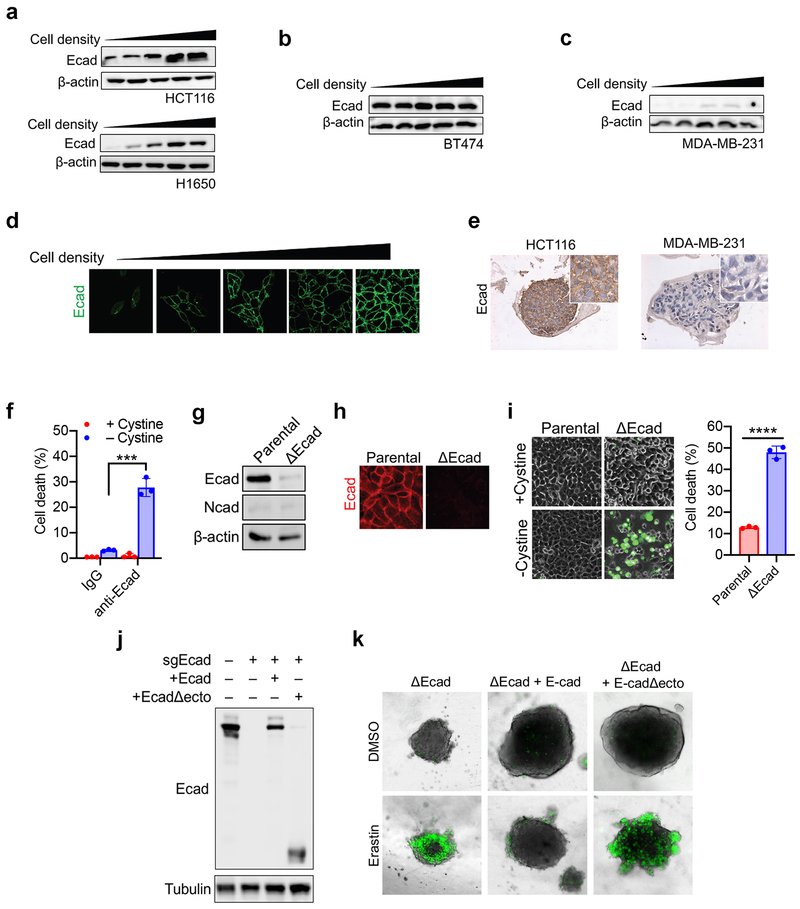
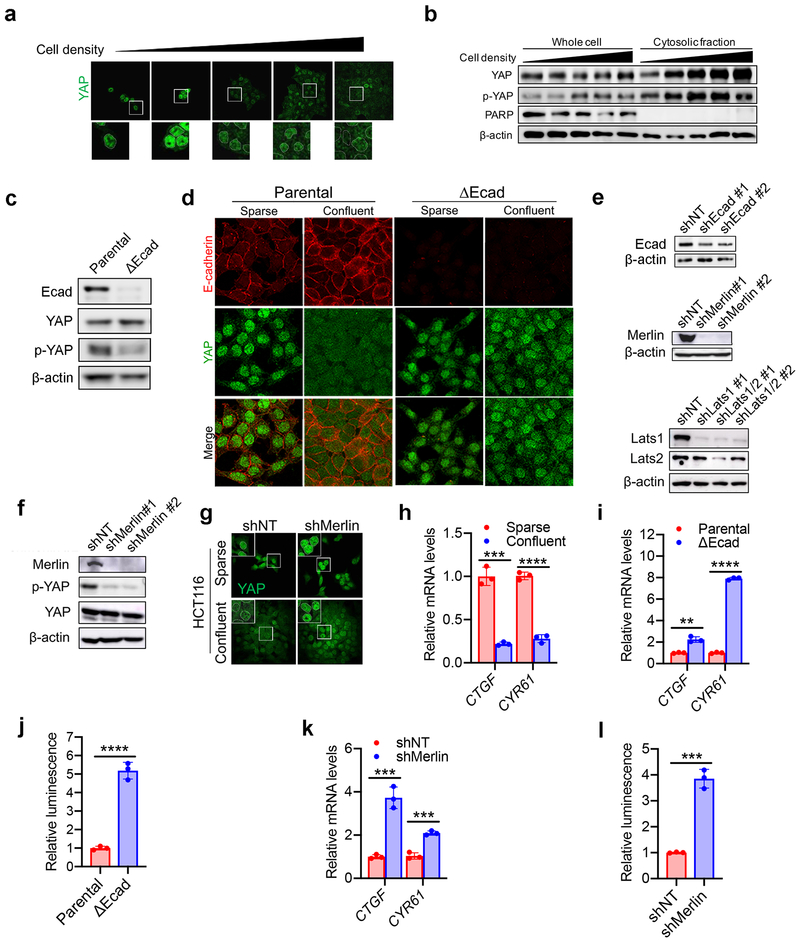
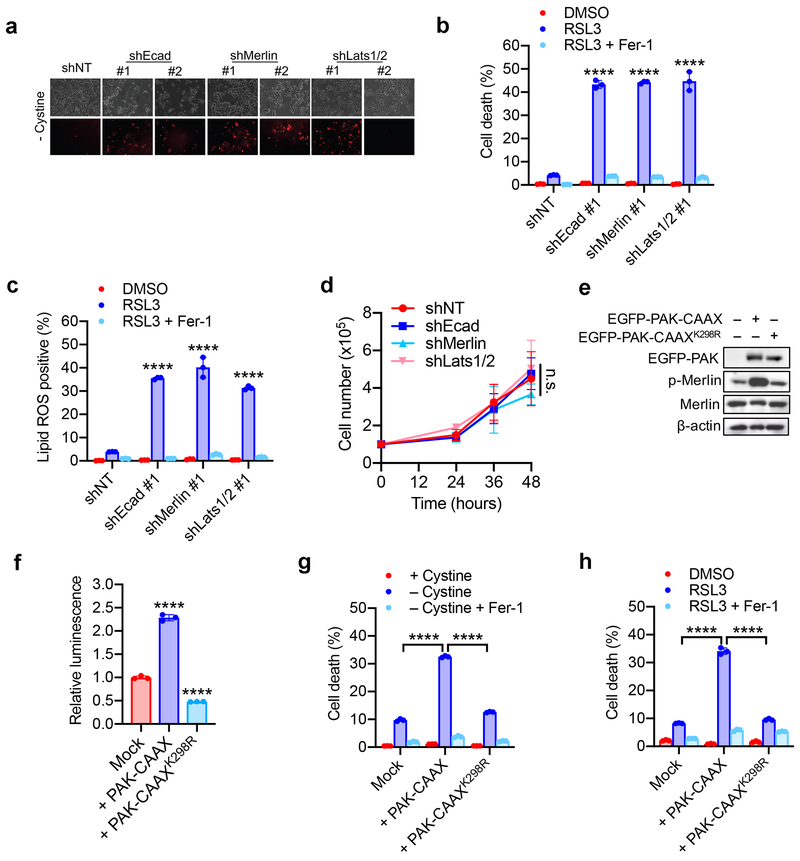
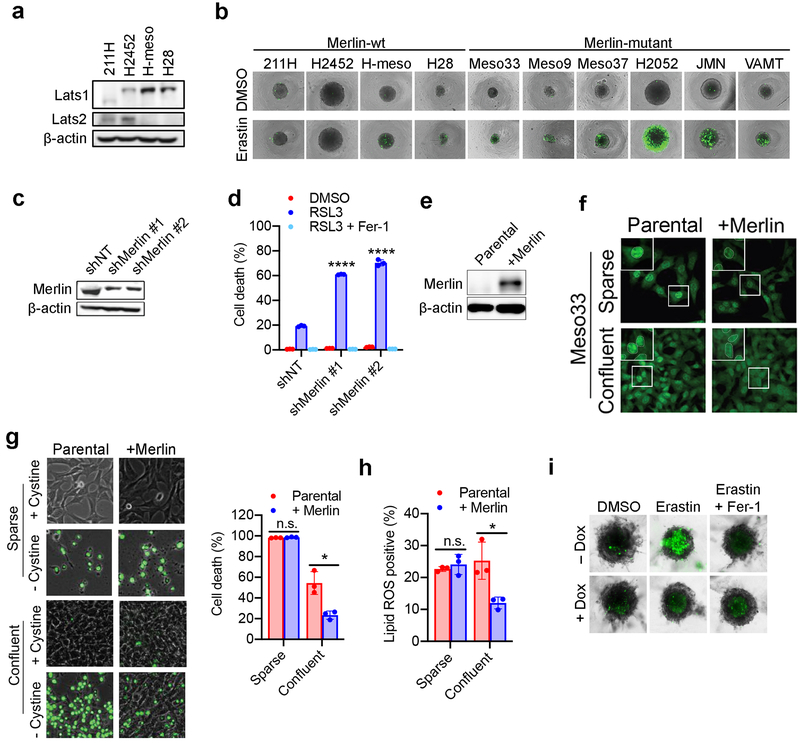
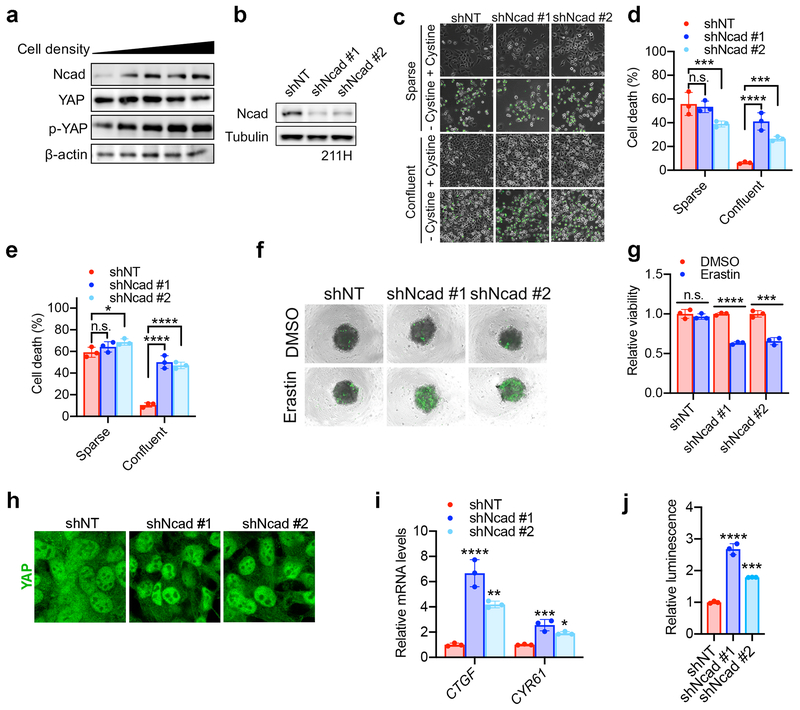
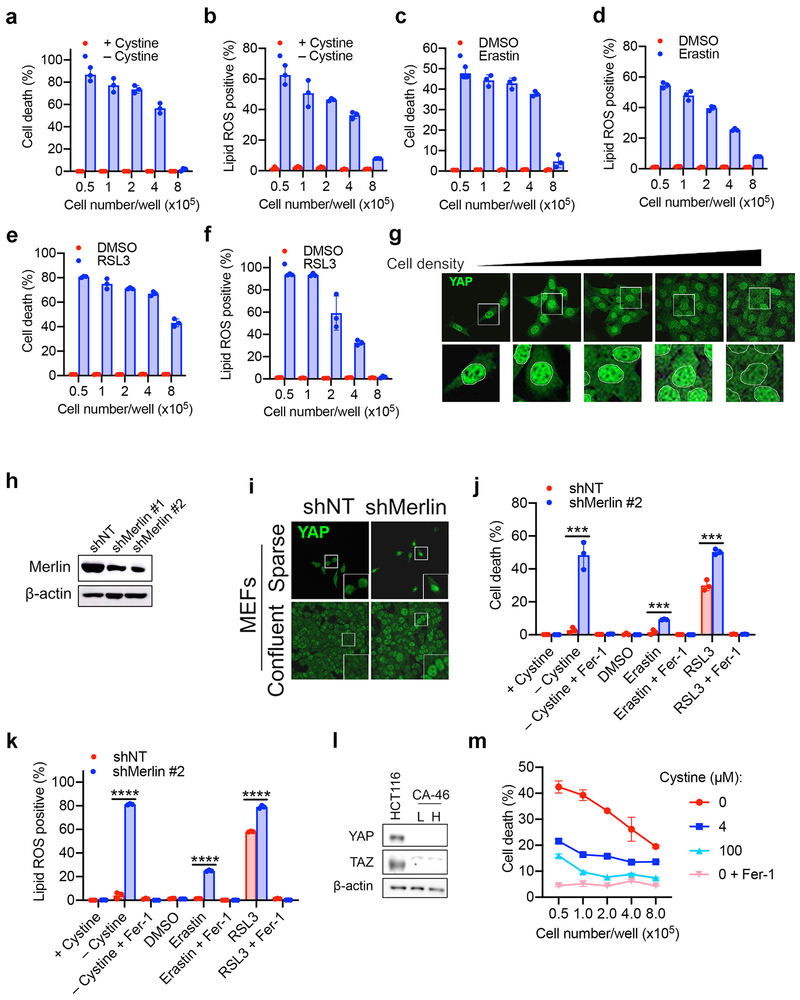
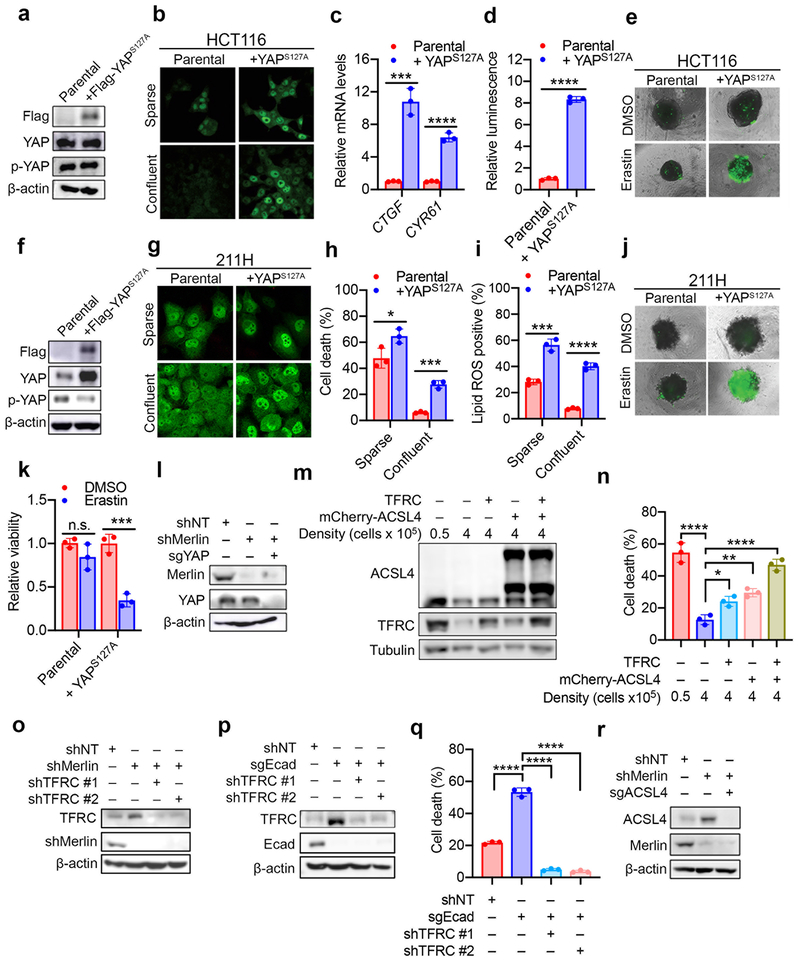
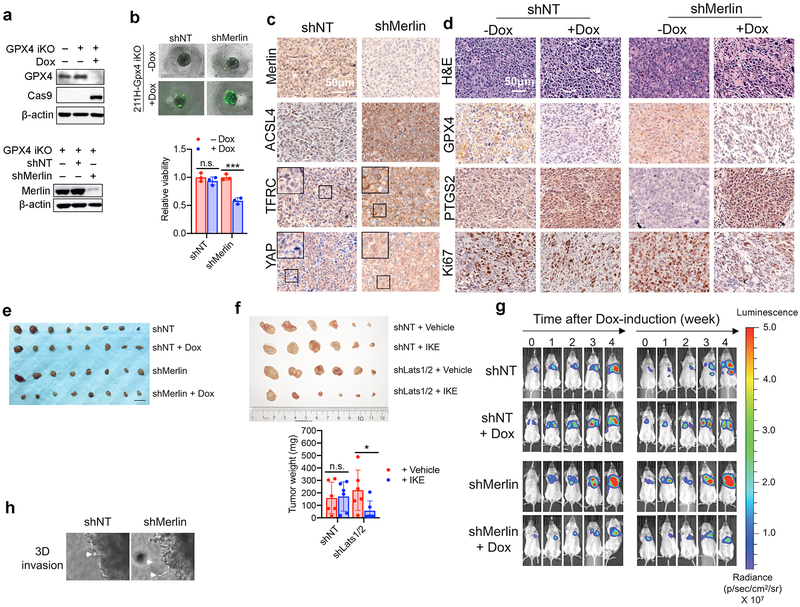
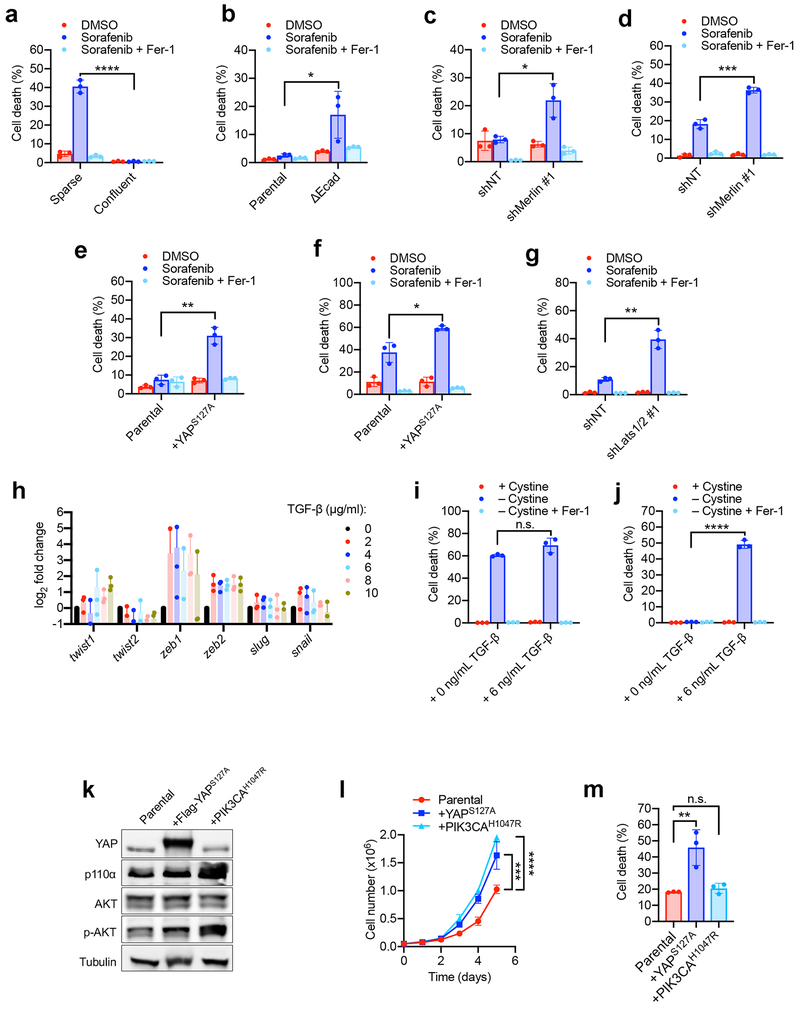
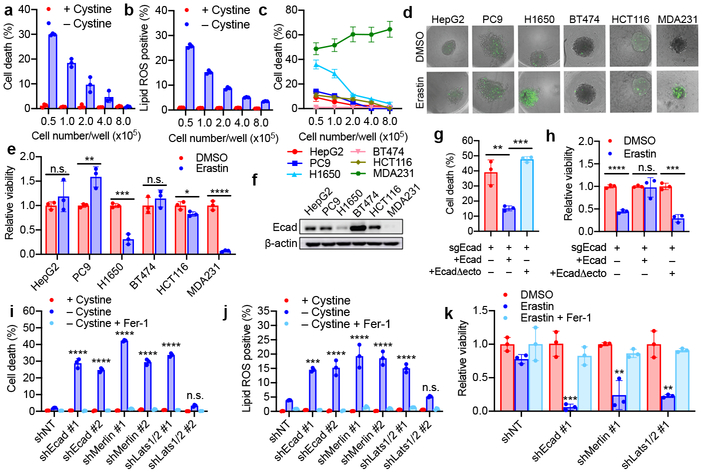
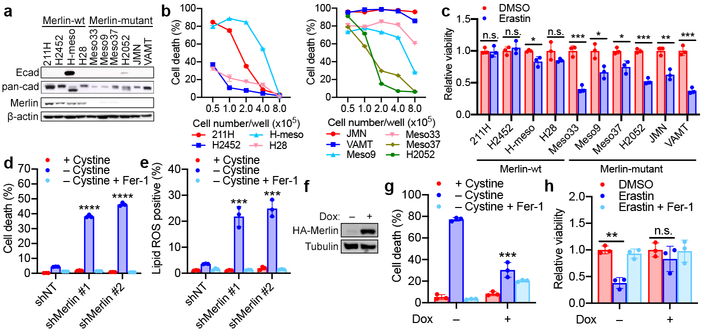
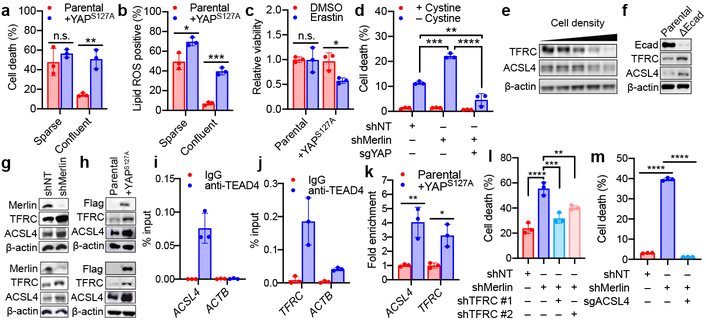
Ferroptosis, a cell death process driven by cellular metabolism and iron-dependent lipid peroxidation, has been implicated in diseases such as ischaemic organ damage and cancer. The enzyme glutathione peroxidase 4 (GPX4) is a central regulator of ferroptosis, and protects cells by neutralizing lipid peroxides, which are by-products of cellular metabolism. The direct inhibition of GPX4, or indirect inhibition by depletion of its substrate glutathione or the building blocks of glutathione (such as cysteine), can trigger ferroptosis. Ferroptosis contributes to the antitumour function of several tumour suppressors such as p53, BAP1 and fumarase. Counterintuitively, mesenchymal cancer cells-which are prone to metastasis, and often resistant to various treatments-are highly susceptible to ferroptosis. Here we show that ferroptosis can be regulated non-cell-autonomously by cadherin-mediated intercellular interactions. In epithelial cells, such interactions mediated by E-cadherin suppress ferroptosis by activating the intracellular NF2 (also known as merlin) and Hippo signalling pathway. Antagonizing this signalling axis allows the proto-oncogenic transcriptional co-activator YAP to promote ferroptosis by upregulating several ferroptosis modulators, including ACSL4 and TFRC. This finding provides mechanistic insights into the observations that cancer cells with mesenchymal or metastatic property are highly sensitive to ferroptosis. Notably, a similar mechanism also modulates ferroptosis in some non-epithelial cells. Finally, genetic inactivation of the tumour suppressor NF2, a frequent tumorigenic event in mesothelioma, rendered cancer cells more sensitive to ferroptosis in an orthotopic mouse model of malignant mesothelioma. Our results demonstrate the role of intercellular interactions and intracellular NF2-YAP signalling in dictating ferroptotic death, and also suggest that malignant mutations in NF2-YAP signalling could predict the responsiveness of cancer cells to future ferroptosis-inducing therapies.
铁死亡是一种由细胞代谢和铁依赖性脂质过氧化驱动的细胞死亡过程,与缺血性器官损伤和癌症等疾病有关。谷胱甘肽过氧化物酶 4(GPX4)是铁死亡的核心调节因子,通过中和细胞代谢的副产物脂质过氧化物来保护细胞。GPX4 的直接抑制或其底物谷胱甘肽或谷胱甘肽构建块(如半胱氨酸)的间接抑制可触发铁死亡。铁死亡有助于几种肿瘤抑制因子(如 p53、BAP1 和延胡索酸酶)的抗肿瘤功能。与直觉相反,间充质癌细胞——易转移,且通常对各种治疗方法有抗性——对铁死亡高度敏感。在这里,我们表明铁死亡可以通过钙黏蛋白介导的细胞间相互作用非自主调节。在上皮细胞中,E-钙黏蛋白介导的这种相互作用通过激活细胞内 NF2(也称为 merlin)和 Hippo 信号通路来抑制铁死亡。拮抗这种信号轴允许原癌基因转录共激活因子 YAP 通过上调几种铁死亡调节剂(包括 ACSL4 和 TFRC)来促进铁死亡。这一发现为以下观察结果提供了机制上的见解:具有间充质或转移特性的癌细胞对铁死亡高度敏感。值得注意的是,类似的机制也调节某些非上皮细胞中的铁死亡。最后,肿瘤抑制因子 NF2 的遗传失活(间皮瘤中的一种常见肿瘤发生事件)使恶性间皮瘤的原位小鼠模型中的癌细胞对铁死亡更敏感。我们的结果表明细胞间相互作用和细胞内 NF2-YAP 信号在决定铁死亡死亡中的作用,并表明 NF2-YAP 信号的恶性突变可能预测癌细胞对未来铁死亡诱导治疗的反应性。