Haryono Mindia, Cho Shu-Ting, Fang Mei-Jane, Chen Ai-Ping, Chou Shu-Jen, Lai Erh-Min, Kuo Chih-Horng
Institute of Plant and Microbial Biology, Academia Sinica, Taipei, Taiwan.
Front Microbiol. 2019 Jul 9;10:1554. doi: 10.3389/fmicb.2019.01554. eCollection 2019.
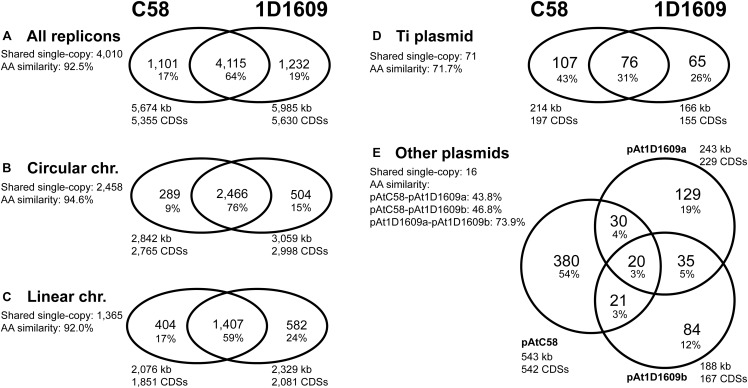
is important in biotechnology due to its ability to transform eukaryotic cells. Although the molecular mechanisms have been studied extensively, previous studies were focused on the model strain C58. Consequently, nearly all of the commonly used strains for biotechnology application were derived from C58 and share similar host ranges. To overcome this limitation, better understanding of the natural genetic variation could provide valuable insights. In this study, we conducted comparative analysis between C58 and 1D1609. These two strains belong to different genomospecies within the species complex and have distinct infectivity profiles. Genome comparisons revealed that each strain has >1,000 unique genes in addition to the 4,115 shared genes. Furthermore, the divergence in gene content and sequences vary among replicons. The circular chromosome is much more conserved compared to the linear chromosome. To identify the genes that may contribute to their differentiation in virulence, we compared the transcriptomes to screen for genes differentially expressed in response to the inducer acetosyringone. Based on the RNA-Seq results with three biological replicates, ∼100 differentially expressed genes were identified in each strain. Intriguingly, homologous genes with the same expression pattern account for <50% of these differentially expressed genes. This finding indicated that phenotypic variation may be partially explained by divergence in expression regulation. In summary, this study characterized the genomic and transcriptomic differences between two representative strains. Moreover, the short list of differentially expressed genes are promising candidates for future characterization, which could improve our understanding of the genetic mechanisms for phenotypic divergence.
由于其转化真核细胞的能力,它在生物技术中很重要。尽管已经对分子机制进行了广泛研究,但先前的研究集中在模式菌株C58上。因此,几乎所有用于生物技术应用的常用菌株都源自C58,并且具有相似的宿主范围。为了克服这一限制,更好地了解自然遗传变异可以提供有价值的见解。在本研究中,我们对C58和1D1609进行了比较分析。这两个菌株属于物种复合体中的不同基因组种,并且具有不同的感染性特征。基因组比较显示,除了4115个共享基因外,每个菌株还有超过1000个独特基因。此外,基因含量和序列的差异在复制子之间有所不同。与线性染色体相比,环状染色体更为保守。为了鉴定可能导致它们毒力分化的基因,我们比较了转录组以筛选响应诱导剂乙酰丁香酮而差异表达的基因。基于三个生物学重复的RNA-Seq结果,在每个菌株中鉴定出约100个差异表达基因。有趣的是,具有相同表达模式的同源基因占这些差异表达基因的不到50%。这一发现表明,表型变异可能部分由表达调控的差异来解释。总之,本研究表征了两个代表性菌株之间的基因组和转录组差异。此外,差异表达基因的简短列表是未来表征的有希望的候选者,这可以提高我们对表型差异遗传机制的理解。