Department of Neurology, Wake Forest School of Medicine, Winston-Salem, North Carolina, USA.
Department of Biostatistical Sciences, Wake Forest School of Medicine, Winston-Salem, North Carolina, USA.
Mov Disord. 2019 Oct;34(10):1528-1536. doi: 10.1002/mds.27801. Epub 2019 Jul 30.
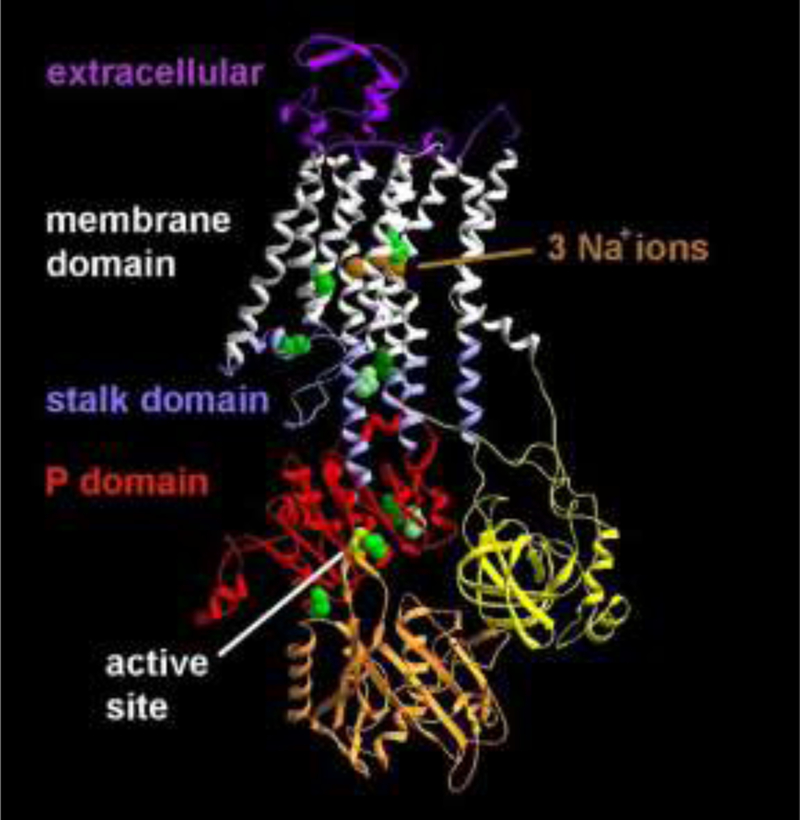
Rapid-onset dystonia-parkinsonism (RDP) is caused by mutations in the ATP1A3 gene, which codes for the α-3 subunit of the Na /K ATPase. It has been characterized by rapid-onset bulbar dysfunction, limb dystonia, bradykinesia, and a rostrocaudal spatial gradient of expression, usually after a physiologic trigger. We reexamined whether these features were in fact characteristic.
We characterized phenotypic variation within a cohort of 50 ATP1A3 mutation-positive individuals (carriers) and 44 mutation-negative family members (noncarriers). Potential participants were gathered through referral for clinical suspicion of RDP or alternating hemiplegia of childhood. Inclusion criteria were having a ATP1A3 mutation or being a family member of such an individual.
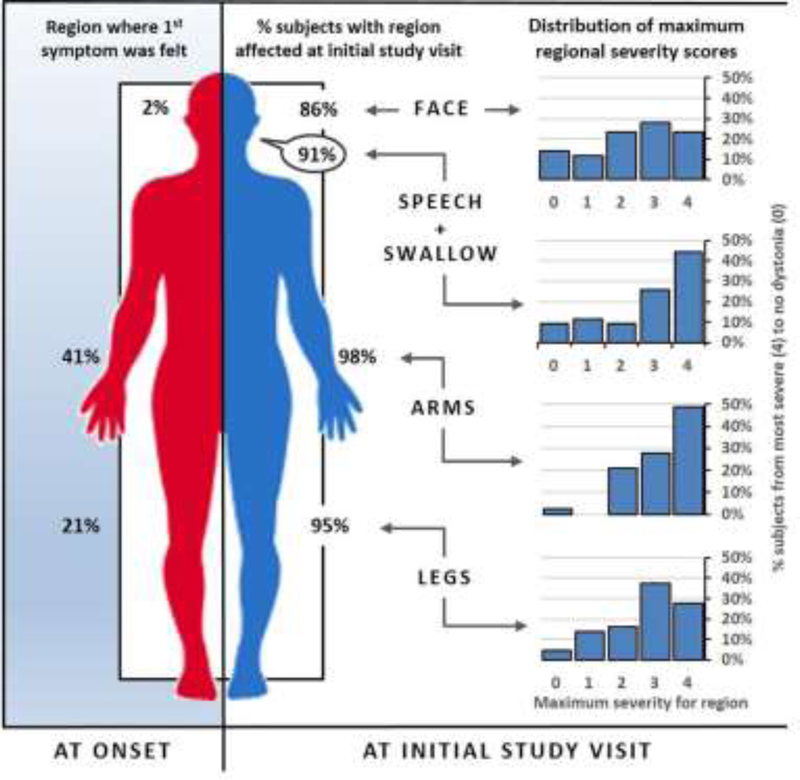
We found RDP is underdiagnosed if only "characteristic" patients are tested. Rapid onset and bulbar predominance were not universally present in carriers. Among those with at least mild symptoms of dystonia, rostrocaudal severity gradient was rare (7%). Symptoms began focally but progressed to be generalized (51%) or multifocal (49%). Arm (41%) onset was most common. Arms and voice were typically most severely affected (48% and 44%, respectively). Triggers preceded onset in 77% of the participants. Rapid onset, dystonia, parkinsonism, bulbar symptoms, headaches, seizures, frontal impairment, and a history of mood disorder and a history of psychosis were more common in carriers. Approximately half of the proband mutations occurred de novo (56%).
Our findings suggest that patients should not be excluded from ATP1A3 testing because of slow onset, limb onset, absent family history, or onset in middle adulthood. RDP should be strongly considered in the differential for any bulbar dystonia. © 2019 International Parkinson and Movement Disorder Society.
快速发作的肌张力障碍-帕金森病(RDP)是由 ATP1A3 基因突变引起的,该基因编码钠/钾 ATP 酶的α-3 亚单位。其特征为快速发作的球部功能障碍、肢体肌张力障碍、运动迟缓,以及表达的头足空间梯度,通常在生理触发后发生。我们重新检查了这些特征是否实际上具有特征性。
我们对 50 名 ATP1A3 突变阳性个体(携带者)和 44 名突变阴性家族成员(非携带者)的队列中的表型变异进行了特征描述。通过临床怀疑 RDP 或交替性偏瘫儿童的转介来收集潜在的参与者。纳入标准为具有 ATP1A3 突变或为该个体的家族成员。
如果仅测试“特征性”患者,我们发现 RDP 的诊断不足。快速发作和球部优势并非在携带者中普遍存在。在至少有轻度肌张力障碍症状的患者中,头足严重程度梯度很少见(7%)。症状开始时呈局灶性,但进展为全身性(51%)或多灶性(49%)。手臂(41%)起病最常见。手臂和声音通常受影响最严重(分别为 48%和 44%)。77%的参与者的症状发作前有触发因素。快速发作、肌张力障碍、帕金森病、球部症状、头痛、癫痫、额叶损伤以及情绪障碍和精神病史在携带者中更为常见。大约一半的先证者突变是从头发生成的(56%)。
我们的研究结果表明,不应因发病缓慢、肢体起病、无家族史或发病于中年而排除患者进行 ATP1A3 检测。任何球部肌张力障碍都应强烈考虑 RDP。