From the Developmental Neurosciences (A.V., K.B., M.A.K., J.H.C.), UCL Great Ormond Street Institute of Child Health, London, UK; Department of Neurology (A.V., L.C., M.A.K., J.H.C.), Great Ormond Street Hospital, London, UK; Department of Clinical Genetics (R.A., M.H.-E.), Guy's and St Thomas' NHS Foundation Trust, Guy's Hospital, London, United Kingdom; Department of Clinical Genetics (S.K., T.T.K., C.A.L.R.), Leiden University Medical Center, The Netherlands; Division of Genetics and Genomics (C.A.B., P.B.A.), the Manton Center for Orphan Disease Research, Boston Children's Hospital, MA; Department of Pediatrics (C.A.B., P.B.A.), Harvard Medical School, Boston, MA; All Wales Medical Genomics Service (A.E.F.), NHS Wales Cardiff and Vale University Health Board, Institute of Medical Genetics, University Hospital of Wales, UK; Division of Cancer and Genetics (A.E.F.), School of Medicine, Cardiff University, UK; Nuffield Department of Clinical Neurosciences (A.H.N., G.K.T.), University of Oxford, UK; Department of Clinical Genetics (E.H., A.M.), Great Ormond Street Hospital, London, UK; Department of Paediatric Neurology (I.H.), Central Manchester University Hospitals NHS Foundation Trust, UK; SW Thames Regional Genetics Service (S.M.), St George's University Hospitals NHS Foundation Trust, UK; Department of Paediatric Neurology (S.R.M.), Ryegate Children's Centre, Sheffield Children's Hospital, United Kingdom; Institute of Genetic Medicine (M.S.), Newcastle Upon Tyne, UK; Clinical Genetics (P.D.T.), Royal Devon & Exeter NHS Foundation Trust, UK; Aneurin Bevan University Health Board (D.D.), Royal Gwent Hospital, Newport, UK; Division of Newborn Medicine (P.B.A.), Boston Children's Hospital, MA; North West Thames Regional Genetics Service (V.C., N.G., S.E.H., J.R.), Northwick Park Hospital, Middlesex, UK; Department of Clinical and Experimental Epilepsy (S.M.S.), UCL Queen Square Institute of Neurology, London, UK; Department of Oncology & Metabolism (M.B.), University of Sheffield, UK; and Sheffield Clinical Genetics Service (M.B.), Sheffield Childrens NHS Foundation Trust, UK.
Neurology. 2022 Oct 4;99(14):e1511-e1526. doi: 10.1212/WNL.0000000000200927. Epub 2022 Jul 18.
is associated with a broad spectrum of predominantly neurologic disorders, which continues to expand beyond the initially defined phenotypes of alternating hemiplegia of childhood, rapid-onset dystonia parkinsonism, and cerebellar ataxia, areflexia, pes cavus, optic atrophy, sensorineural hearing loss syndrome. This phenotypic variability makes it challenging to assess the pathogenicity of an variant found in an undiagnosed patient. We describe the phenotypic features of individuals carrying a pathogenic/likely pathogenic variant and perform a literature review of all variants published thus far in association with human neurologic disease. Our aim is to demonstrate the heterogeneous clinical spectrum of the gene and look for phenotypic overlap between patients that will streamline the diagnostic process.
Undiagnosed individuals with variants were identified within the cohort of the Deciphering Developmental Disorders study with additional cases contributed by collaborators internationally. Detailed clinical data were collected with consent through a questionnaire completed by the referring clinicians. PubMed was searched for publications containing the term "ATP1A3" from 2004 to 2021.
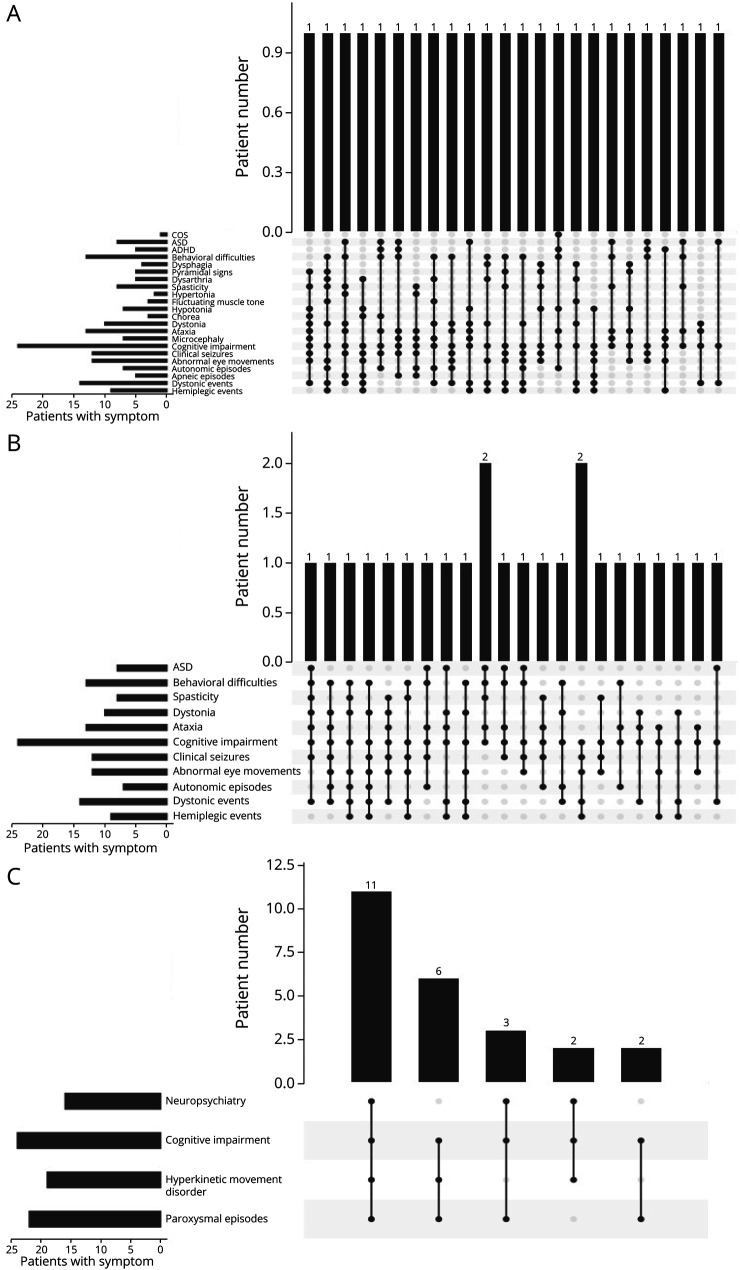
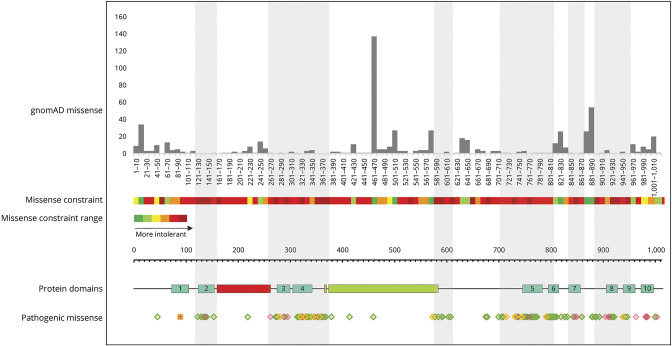
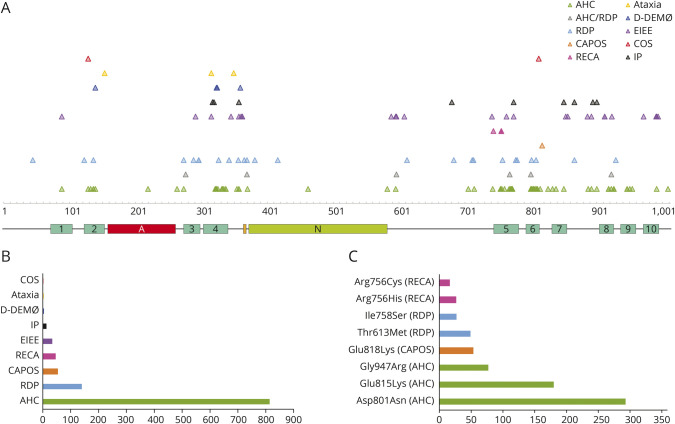
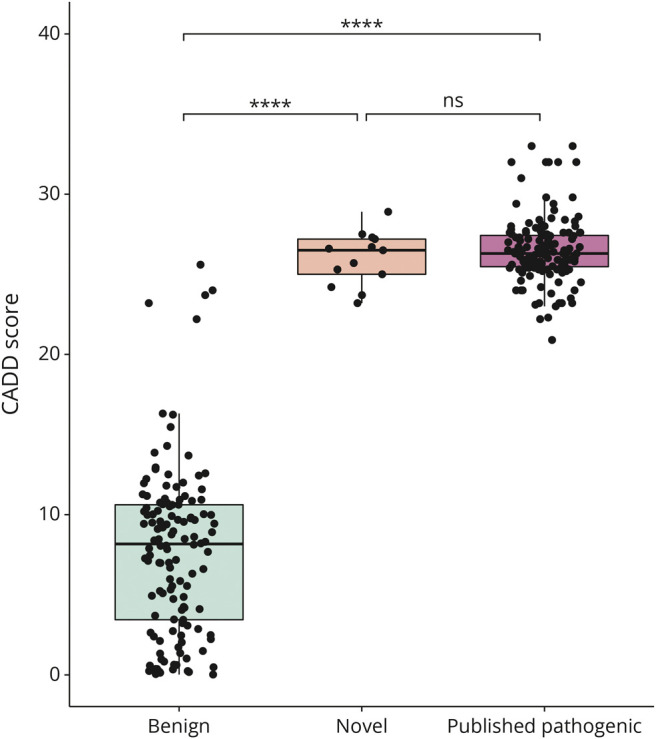
Twenty-four individuals with a previously undiagnosed neurologic phenotype were found to carry 21 variants. Eight variants have been previously published. Patients experienced on average 2-3 different types of paroxysmal events. Permanent neurologic features were common including microcephaly (7; 29%), ataxia (13; 54%), dystonia (10; 42%), and hypotonia (7; 29%). All patients had cognitive impairment. Neuropsychiatric diagnoses were reported in 16 (66.6%) individuals. Phenotypes were extremely varied, and most individuals did not fit clinical criteria for previously published phenotypes. On review of the literature, 1,108 individuals have been reported carrying 168 different variants. The most common variants are associated with well-defined phenotypes, while more rare variants often result in very rare symptom correlations, such as are seen in our study. Combined Annotation-Dependent Depletion (CADD) scores of pathogenic and likely pathogenic variants were significantly higher and variants clustered within 6 regions of constraint.
Our study shows that looking for a combination of paroxysmal events, hyperkinesia, neuropsychiatric symptoms, and cognitive impairment and evaluating the CADD score and variant location can help identify an -related condition, rather than applying diagnostic criteria alone.
ATP1A3 基因突变与广泛的主要为神经障碍相关,这些疾病的表型不断扩展,超出了最初定义的儿童交替性偏瘫、快速进展性肌张力障碍帕金森病、小脑共济失调、反射消失、高弓足、视神经萎缩、感觉神经性听力损失综合征等表型。这种表型的可变性使得评估在未确诊患者中发现的变异的致病性变得具有挑战性。我们描述了携带致病性/可能致病性 ATP1A3 变异的个体的表型特征,并对迄今为止所有与人类神经疾病相关的 ATP1A3 变异进行了文献综述。我们的目的是展示该基因的异质性临床谱,并寻找表型重叠的患者,从而简化诊断过程。
通过 Deciphering Developmental Disorders 研究中的队列鉴定出携带 ATP1A3 变异的未确诊个体,并通过国际合作者提供额外的病例。通过参考医生填写的问卷,在获得同意的情况下收集详细的临床数据。在 2004 年至 2021 年期间,在 PubMed 上以术语“ATP1A3”搜索出版物。
发现 24 名患有先前未确诊的神经表型的个体携带 21 种变异。其中 8 种变异之前已发表。患者平均经历 2-3 种不同类型的阵发性事件。永久性神经特征很常见,包括小头畸形(7;29%)、共济失调(13;54%)、肌张力障碍(10;42%)和低张力(7;29%)。所有患者均存在认知障碍。16 名(66.6%)患者报告存在神经精神诊断。表型极为多样,大多数患者不符合先前发表的表型的临床标准。在对文献的回顾中,共报道了 1108 名个体携带 168 种不同的 ATP1A3 变异。最常见的变异与明确的表型相关,而更罕见的变异通常与非常罕见的症状相关,如我们的研究中所见。致病性和可能致病性变异的综合注释依赖性耗竭(CADD)评分显著较高,并且变异集中在 6 个约束区域内。
我们的研究表明,寻找阵发性事件、多动、神经精神症状和认知障碍的组合,并评估 CADD 评分和变异位置有助于确定与 ATP1A3 相关的疾病,而不是单独应用诊断标准。