Institute of Environmental Science & Research (ESR),Porirua,New Zealand.
Epidemiol Infect. 2019 Jan;147:e186. doi: 10.1017/S0950268819000773.
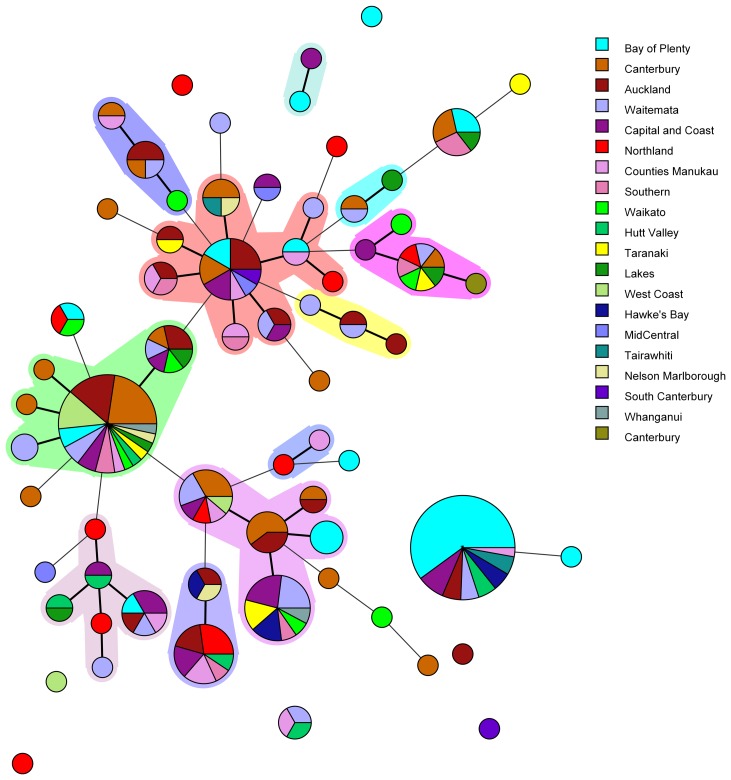
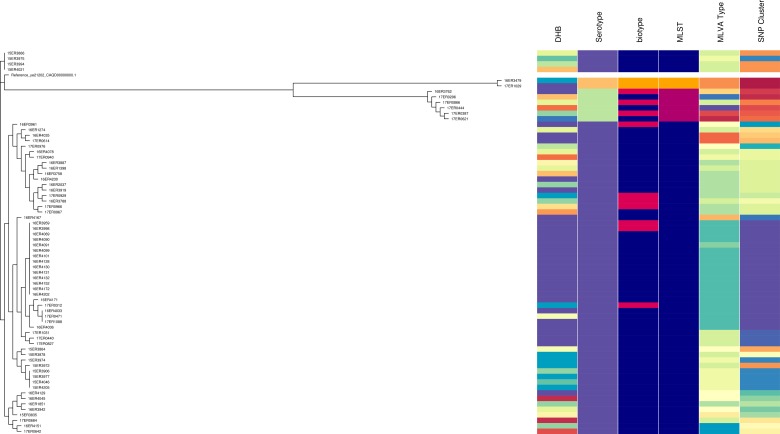
Incidence of human yersiniosis in New Zealand has increased between 2013 and 2017. For surveillance and outbreak investigations it is essential that an appropriate level of discrimination between pathogenic Yersinia enterocolitica isolates is provided, in order to support epidemiological linking of connected cases. Subtyping of 227 Y. enterocolitica isolates was performed using a range of different typing methods, including biotyping, serotyping and seven loci multiple-locus variable-number tandem-repeat analysis (MLVA). In addition, core genome single-nucleotide polymorphism (core SNP) analysis and multi-locus sequence typing were performed on a subset of 69 isolates. Sixty-seven different MLVA types were identified. One MLVA profile was associated with an outbreak in the Bay of Plenty region, supported by epidemiological data. Core SNP analysis showed that all the outbreak-related isolates clustered together. The subtyping and epidemiological evidence suggests that the outbreak of yersiniosis in the Bay of Plenty region between October and December 2016 could be attributed to a point source. However, subtyping results further suggest that the same clone was isolated from several regions between August 2016 and March 2017. Core SNP analysis and MLVA typing failed to differentiate between Y. enterocolitica biotype 2 and biotype 3. For this reason, we propose that these biotypes should be reported as a single type namely: Y. enterocolitica biotype 2/3 and that the serotype should be prioritised as an indicator of prevalence.
2013 年至 2017 年间,新西兰人类耶尔森菌病的发病率有所增加。为了进行监测和暴发调查,必须提供致病性小肠结肠炎耶尔森菌分离株之间适当水平的区分,以支持相关病例的流行病学联系。使用一系列不同的分型方法对 227 株小肠结肠炎耶尔森菌进行了分型,包括生物分型、血清分型和 7 个基因座多位点可变数串联重复分析(MLVA)。此外,对 69 株分离株的核心基因组单核苷酸多态性(core SNP)分析和多位点序列分型进行了分析。确定了 67 种不同的 MLVA 类型。一种 MLVA 图谱与 2016 年 10 月至 12 月在丰盛湾地区暴发的疫情有关,这一结果得到了流行病学数据的支持。核心 SNP 分析表明,所有与暴发相关的分离株聚集在一起。分型和流行病学证据表明,2016 年 10 月至 12 月丰盛湾地区的耶尔森菌病暴发可能归因于一个单一来源。然而,分型结果进一步表明,同一克隆在 2016 年 8 月至 2017 年 3 月期间从几个地区分离出来。核心 SNP 分析和 MLVA 分型无法区分小肠结肠炎耶尔森菌生物型 2 和生物型 3。因此,我们建议将这两种生物型报告为一种类型,即:小肠结肠炎耶尔森菌生物型 2/3,血清型应作为流行的指标。