Department of Computer Science, City University of Hong Kong, Hong Kong 999077, China.
Department of Respiratory Diseases, Shenzhen Children's Hospital, Shenzhen 518026, China.
Gigascience. 2019 Aug 1;8(8). doi: 10.1093/gigascience/giz093.
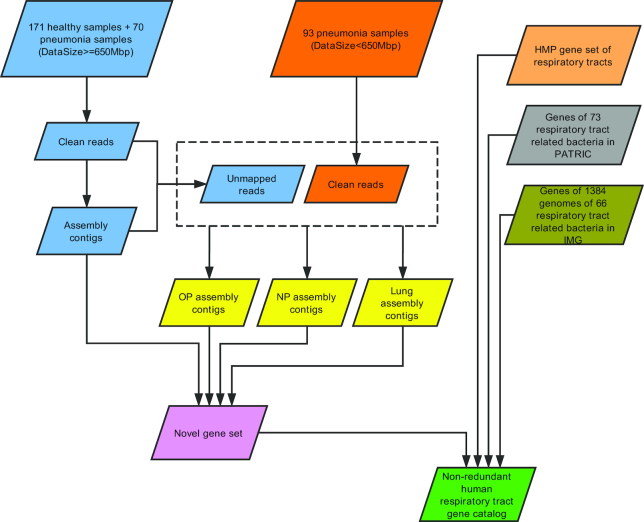
The imbalanced respiratory microbiota observed in pneumonia causes high morbidity and mortality in childhood. Respiratory metagenomic analysis demands a comprehensive microbial gene catalogue, which will significantly advance our understanding of host-microorganism interactions.
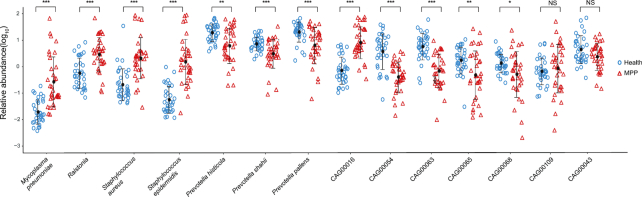
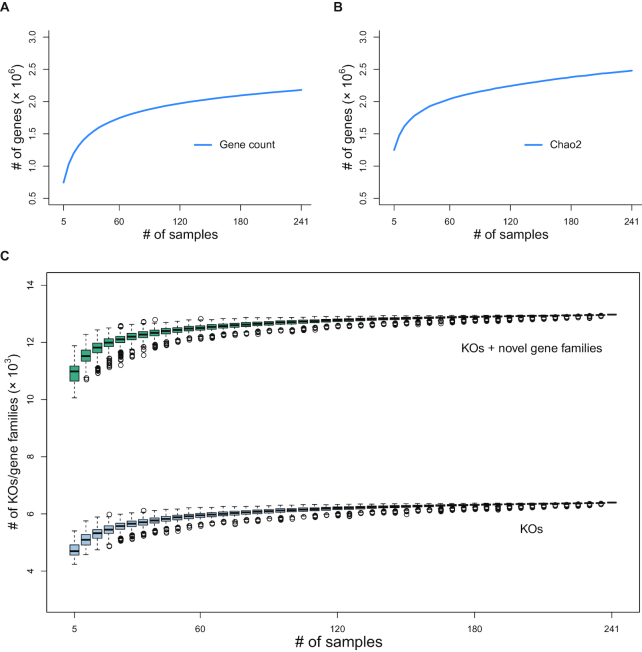
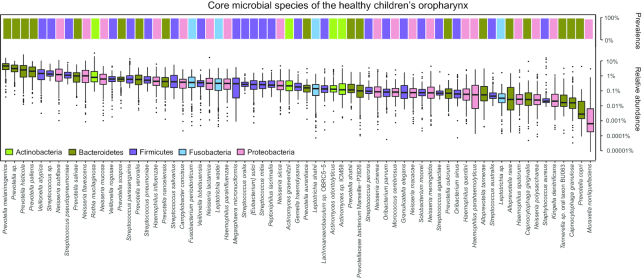
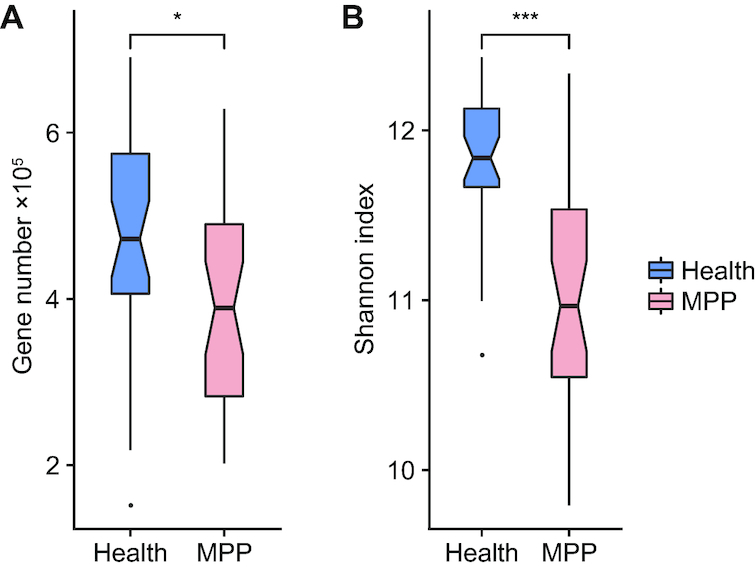
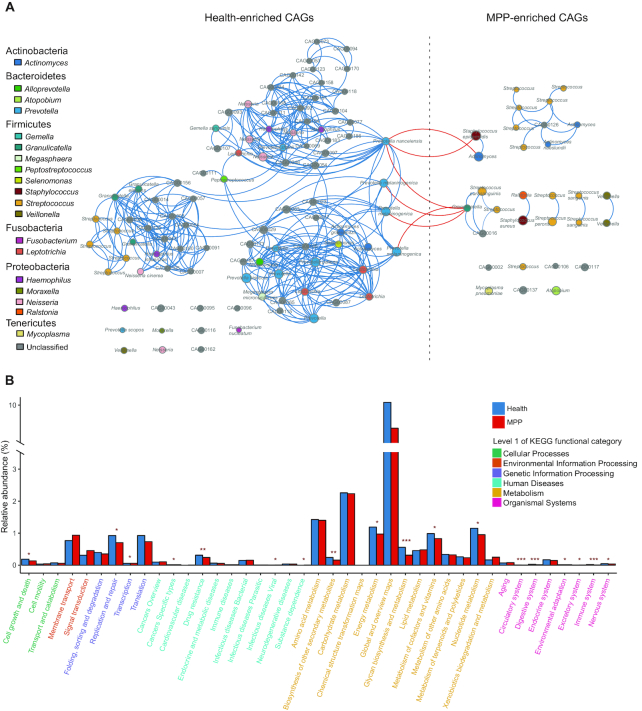
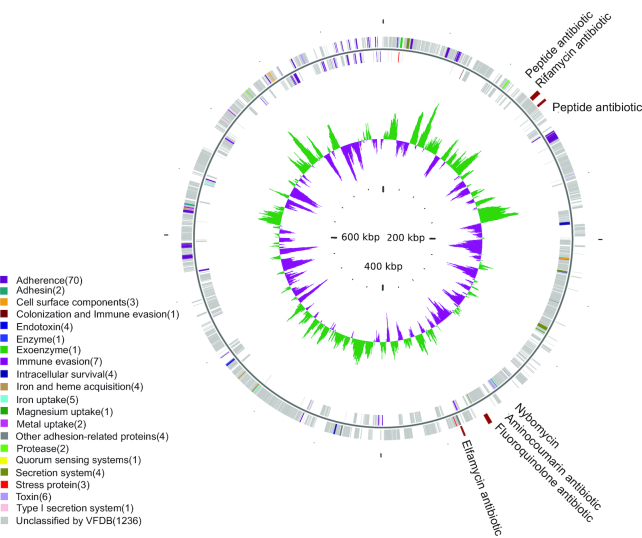
We collected 334 respiratory microbial samples from 171 healthy children and 76 children with pneumonia. The respiratory microbial gene catalogue we established comprised 2.25 million non-redundant microbial genes, covering 90.52% of prevalent genes. The major oropharyngeal microbial species found in healthy children were Prevotella and Streptococcus. In children with Mycoplasma pneumoniae pneumonia (MPP), oropharyngeal microbial diversity and associated gene numbers decreased compared with those of healthy children. The concurrence network of oropharyngeal microorganisms in patients predominantly featured Staphylococcus spp. and M. pneumoniae. Functional orthologues, which are associated with the metabolism of various lipids, membrane transport, and signal transduction, accumulated in the oropharyngeal microbiome of children with pneumonia. Several antibiotic resistance genes and virulence factor genes were identified in the genomes of M. pneumoniae and 13 other microorganisms reconstructed via metagenomic data. Although the common macrolide/β-lactam resistance genes were not identified in the assembled M. pneumoniae genome, a single-nucleotide polymorphism (A2063G) related to macrolide resistance was identified in a 23S ribosomal RNA gene.
The results of this study will facilitate exploration of unknown microbial components and host-microorganism interactions in studies of the respiratory microbiome. They will also yield further insights into the microbial aetiology of MPP.
肺炎中观察到的呼吸微生物群落失衡导致儿童发病率和死亡率居高不下。呼吸宏基因组分析需要一个全面的微生物基因目录,这将极大地促进我们对宿主-微生物相互作用的理解。
我们从 171 名健康儿童和 76 名肺炎儿童中收集了 334 个呼吸道微生物样本。我们建立的呼吸道微生物基因目录包含 225 万个非冗余微生物基因,涵盖了 90.52%的常见基因。在健康儿童中,主要的口咽微生物物种为普雷沃菌属和链球菌属。在肺炎支原体肺炎(MPP)患儿中,与健康儿童相比,口咽微生物多样性和相关基因数量减少。患者口咽微生物的共现网络主要以葡萄球菌属和肺炎支原体为主。与各种脂质代谢、膜转运和信号转导相关的功能直系同源物在肺炎患儿的口咽微生物组中积累。在通过宏基因组数据重建的肺炎支原体和其他 13 种微生物的基因组中,鉴定出了几种抗生素耐药基因和毒力因子基因。虽然在组装的肺炎支原体基因组中未发现常见的大环内酯类/β-内酰胺类耐药基因,但在 23S 核糖体 RNA 基因中发现了一个与大环内酯类耐药相关的单核苷酸多态性(A2063G)。
本研究的结果将有助于探索呼吸微生物组中未知的微生物成分和宿主-微生物相互作用,并进一步了解 MPP 的微生物病因。