Computational Biology Department, School of Computer Science, Carnegie Mellon University, Pittsburgh, PA, USA.
Genome Biol. 2018 Feb 7;19(1):16. doi: 10.1186/s13059-018-1388-2.
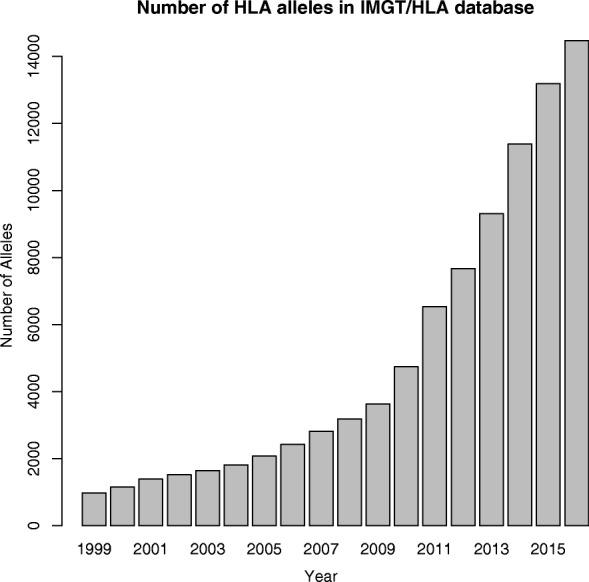
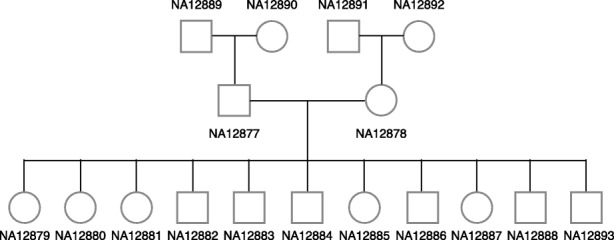
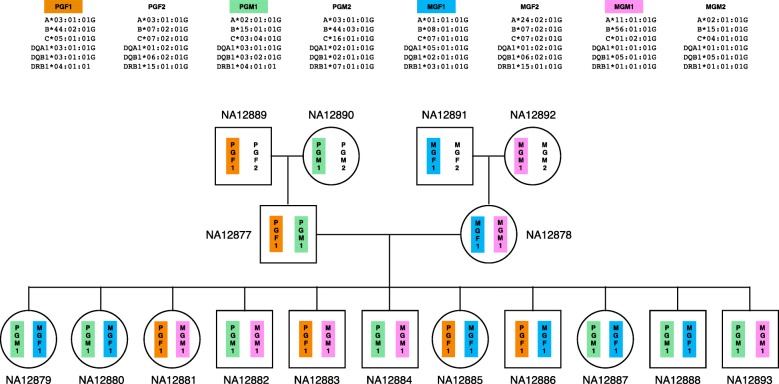
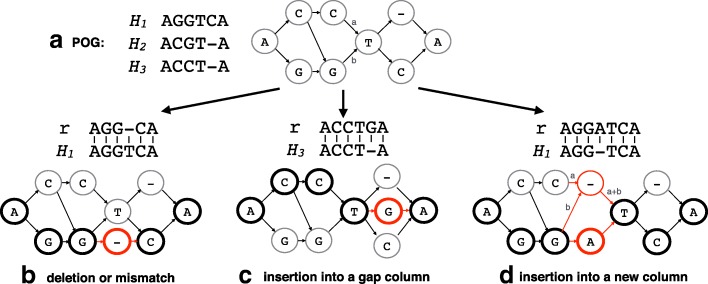
Accurate typing of human leukocyte antigen (HLA) is important because HLA genes play important roles in immune responses and disease genesis. Previously available computational methods are database-matching approaches and their outputs are inherently limited by the completeness of already known types, making them unsuitable for discovery of novel alleles. We have developed a graph-guided assembly technique for classical HLA genes, which can construct allele sequences given high-coverage whole-genome sequencing data. Our method delivers highly accurate HLA typing, comparable to the current state-of-the-art methods. Using various data, we also demonstrate that our method can type novel alleles.
准确的人类白细胞抗原(HLA)分型非常重要,因为 HLA 基因在免疫反应和疾病发生中起着重要作用。以前可用的计算方法是数据库匹配方法,其输出本质上受到已知类型完整性的限制,因此不适合发现新的等位基因。我们开发了一种用于经典 HLA 基因的图引导组装技术,该技术可以根据高覆盖率的全基因组测序数据构建等位基因序列。我们的方法提供了高度准确的 HLA 分型,可与当前最先进的方法相媲美。使用各种数据,我们还证明了我们的方法可以对新的等位基因进行分型。