Zakham F, Laurent S, Esteves Carreira A L, Corbaz A, Bertelli C, Masserey E, Nicod L, Greub G, Jaton K, Mazza-Stalder J, Opota O
Institute of Microbiology, University of Lausanne and University Hospital of Lausanne, Lausanne Switzerland.
Department of Pneumology, University of Lausanne and University Hospital of Lausanne, Lausanne Switzerland.
New Microbes New Infect. 2019 Jun 29;31:100582. doi: 10.1016/j.nmni.2019.100582. eCollection 2019 Sep.
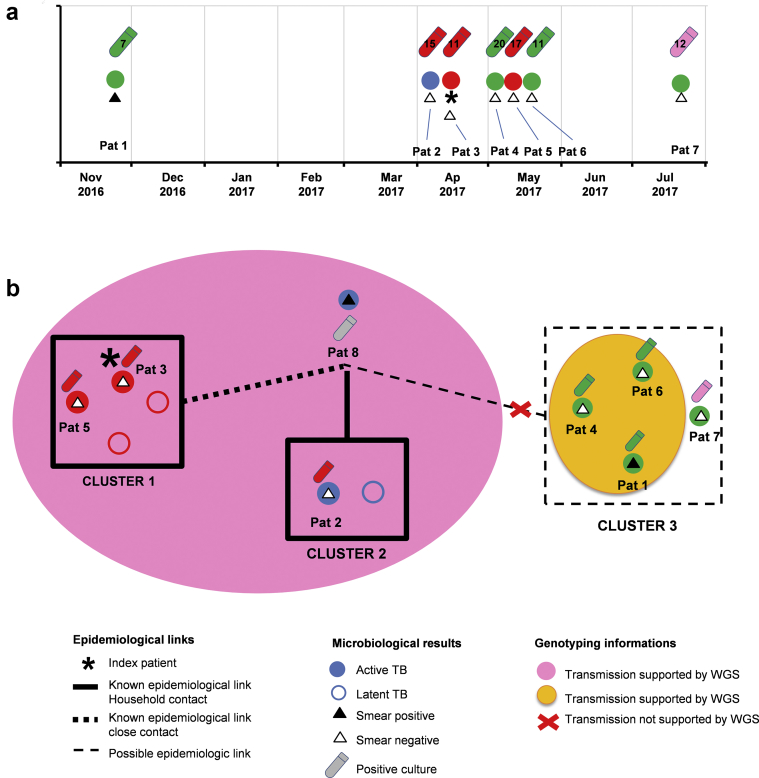
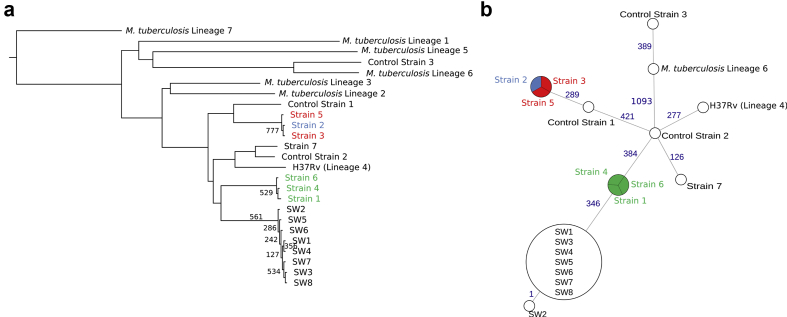
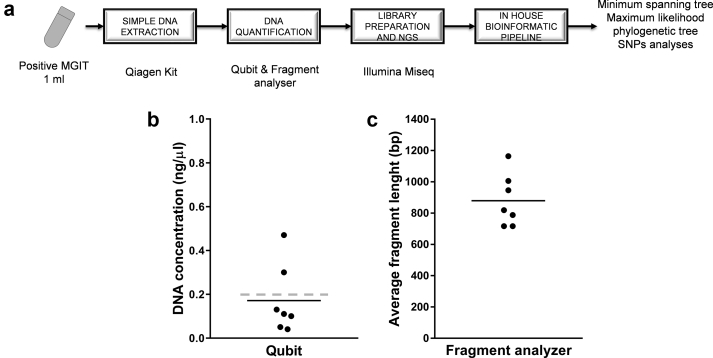
Contact investigations following the diagnosis of active tuberculosis (TB) are paramount for the control of the disease. Epidemiological data are very powerful for contact tracing but might be delayed and/or difficult to integrate, especially in the setting of multiple contact-tracing investigations. The aim of this study was to address the added-value of whole-genome sequencing (WGS) to routine local TB surveillance systems. From November 2016 to July 2017, the local TB surveillance system identified three clusters that could constitute a unique larger outbreak. Epidemiological and clinical information were integrated with WGS genotyping data of strains obtained using a simple DNA extraction method coupled with sequencing using an Illumina MiSeq platform and an in-house bioinformatics pipeline for single nucleotide polymorphism (SNP) analysis. Epidemiological investigations identified three putative TB clusters potentially interrelated including eight patients with active TB. Seven isolates were available and analysed by WGS. Using a 5-SNP threshold to define recent transmission, WGS-based genotyping supported the occurrence of the three clusters as well as a link between clusters 1 and 2 (SNP ≤1), constituting a larger outbreak. This outbreak was clearly delineated by refuting a potential link with the third cluster (SNP >500). Genotyping data did not support the belonging of patient 7 to any studied cluster. This study illustrates the usefulness of WGS genotyping for routine TB surveillance systems in local communities to rapidly confirm or disprove epidemiological hypotheses and delineate TB clusters, especially in the context of multiple contact-tracing investigations.
活动性结核病(TB)诊断后的接触者调查对于该疾病的控制至关重要。流行病学数据在接触者追踪方面非常有力,但可能会延迟和/或难以整合,尤其是在进行多次接触者追踪调查的情况下。本研究的目的是探讨全基因组测序(WGS)对常规地方结核病监测系统的附加价值。2016年11月至2017年7月,地方结核病监测系统识别出三个可能构成一次独特更大规模疫情的聚集性病例。将流行病学和临床信息与通过简单DNA提取方法获得的菌株的WGS基因分型数据相结合,该方法结合了使用Illumina MiSeq平台进行的测序以及用于单核苷酸多态性(SNP)分析的内部生物信息学流程。流行病学调查确定了三个可能相互关联的假定结核病聚集性病例,包括八名活动性结核病患者。获得了七株分离株并通过WGS进行分析。使用5个SNP的阈值来定义近期传播,基于WGS的基因分型支持了这三个聚集性病例的存在以及聚集性病例1和2之间的联系(SNP≤1),构成了一次更大规模的疫情。通过排除与第三个聚集性病例的潜在联系(SNP>500),明确划定了这次疫情。基因分型数据不支持患者7属于任何所研究的聚集性病例。本研究说明了WGS基因分型对于地方社区常规结核病监测系统快速证实或反驳流行病学假设以及划定结核病聚集性病例的有用性,尤其是在进行多次接触者追踪调查的情况下。