Sivaprakash S, Prakash S, Mohan S, Jose Sujin P
Department of Computational Physics, School of Physics, Madurai Kamaraj University, Madurai, 625 021, Tamil Nadu, India.
Departmentof Physics, VHNSN College, Virudhunagar, 626 001, Tamil Nadu, India.
Heliyon. 2019 Jul 29;5(7):e02149. doi: 10.1016/j.heliyon.2019.e02149. eCollection 2019 Jul.
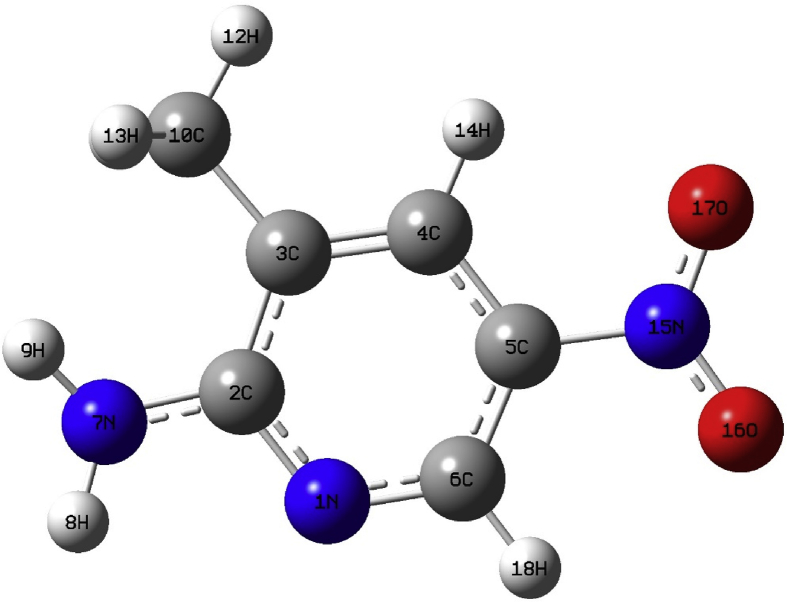
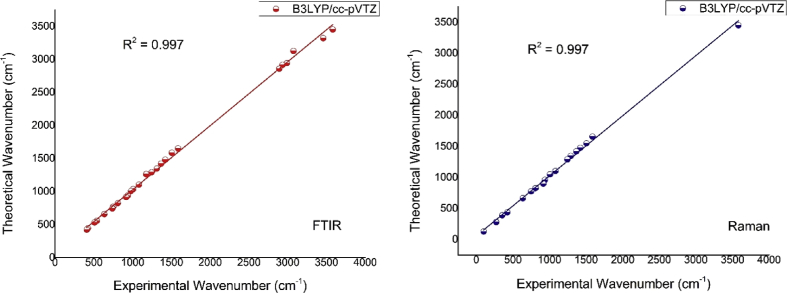
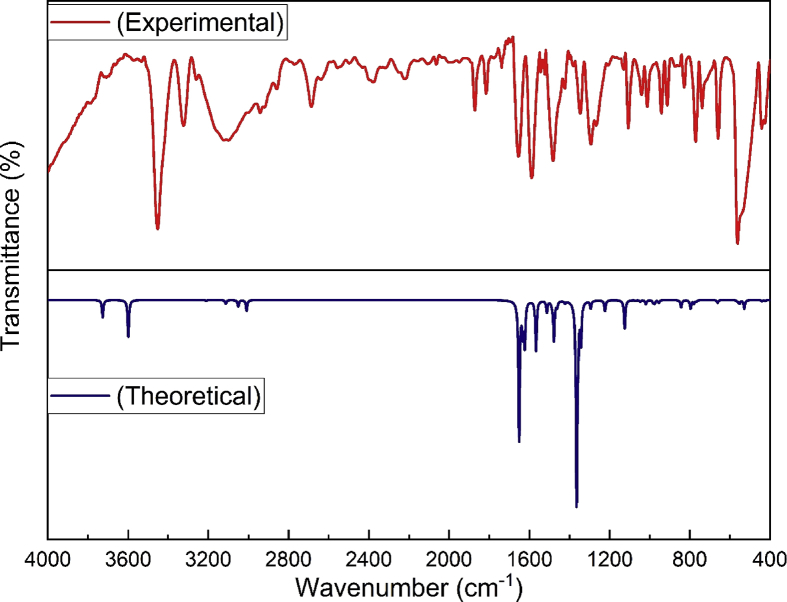
Quantum chemical calculations on energy and molecular structure of 2-amino-3-methyl-5-nitropyridine (2A3M5NP) have been attempted by implementing DFT/B3LYP method using 6-311G (d,p), 6-311G++ (d,p) and cc-pVTZ basis sets. The optimized geometry and the vibrational analysis for energetically most stable configuration, are carried out theoretically by using B3LYP/cc-pVTZ basis set. The computed vibrational frequencies were scaled by using scaling factors and compared with the experimental Fourier Transform Infra-Red (FTIR) solid phase spectrum in the region 4000-400 cm and FT-Raman spectrum in the region 4000-100 cm. The complete vibrational assignments, analysis and correlation of fundamental modes of the compound have been carried out using the potential energy distribution (PED). The intramolecular charge transfer, hyperconjugative interaction of the compound is investigated from natural bonding orbital (NBO) analysis. The UV-Visible spectrum of 2A3M5NP was obtained with ethanol as a solvent. The electronic properties such as HOMO (Highest Occupied Molecular Orbital) and LUMO (Lowest Unoccupied Molecular Orbital) energies are determined by B3LYP/cc-pVTZ basis set. The electronic absorption spectrum of the compound was studied from UV-Visible analysis by using time-dependent density functional theory (TD-DFT). The electron density distribution and chemical reactive sites of 2A3M5NP were analyzed from molecular electrostatic potential (MEP) analysis and frontier molecular orbital (FMO) analysis.
通过使用6-311G(d,p)、6-311G++(d,p)和cc-pVTZ基组实施密度泛函理论/ B3LYP方法,对2-氨基-3-甲基-5-硝基吡啶(2A3M5NP)的能量和分子结构进行了量子化学计算。使用B3LYP/cc-pVTZ基组从理论上对能量最稳定构型进行了优化几何结构和振动分析。计算得到的振动频率通过比例因子进行缩放,并与4000-400 cm区域的实验傅里叶变换红外(FTIR)固相光谱以及4000-100 cm区域的傅里叶变换拉曼(FT-Raman)光谱进行比较。利用势能分布(PED)对该化合物的基本振动模式进行了完整的振动归属、分析和关联。通过自然键轨道(NBO)分析研究了该化合物的分子内电荷转移和超共轭相互作用。以乙醇为溶剂获得了2A3M5NP的紫外可见光谱。通过B3LYP/cc-pVTZ基组确定了诸如最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)能量等电子性质。通过使用含时密度泛函理论(TD-DFT)的紫外可见分析研究了该化合物的电子吸收光谱。通过分子静电势(MEP)分析和前线分子轨道(FMO)分析对2A3M5NP的电子密度分布和化学反应位点进行了分析。