Scheie Eye Institute, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, 19104, USA.
FM Kirby Center for Molecular Ophthalmology, University of Pennsylvania, Stellar-Chance Laboratories, 3rd Floor, 422 Curie Blvd, Philadelphia, PA, 19104, USA.
Sci Rep. 2019 Aug 12;9(1):11664. doi: 10.1038/s41598-019-48087-3.
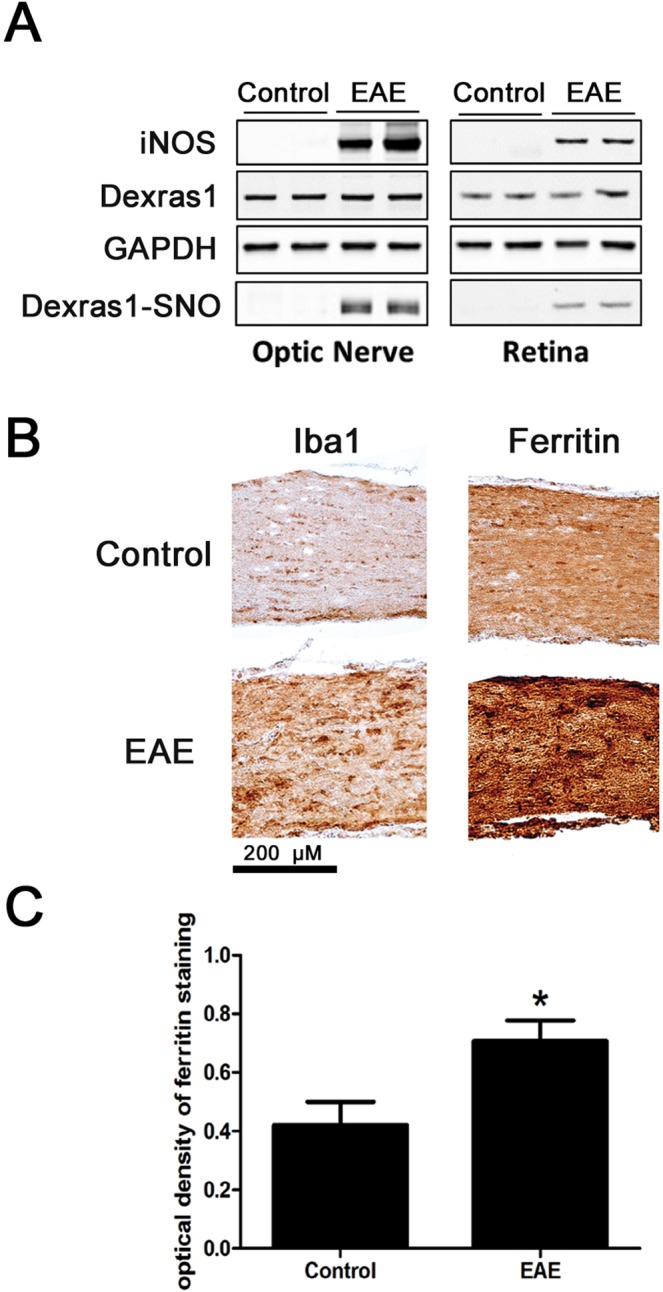
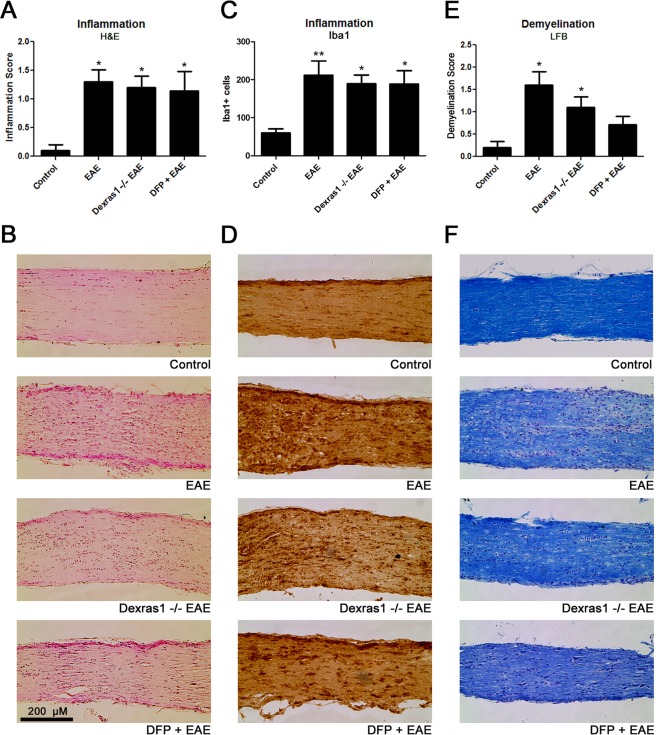
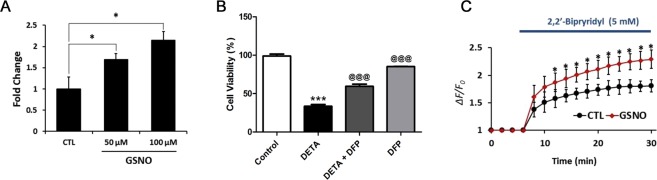
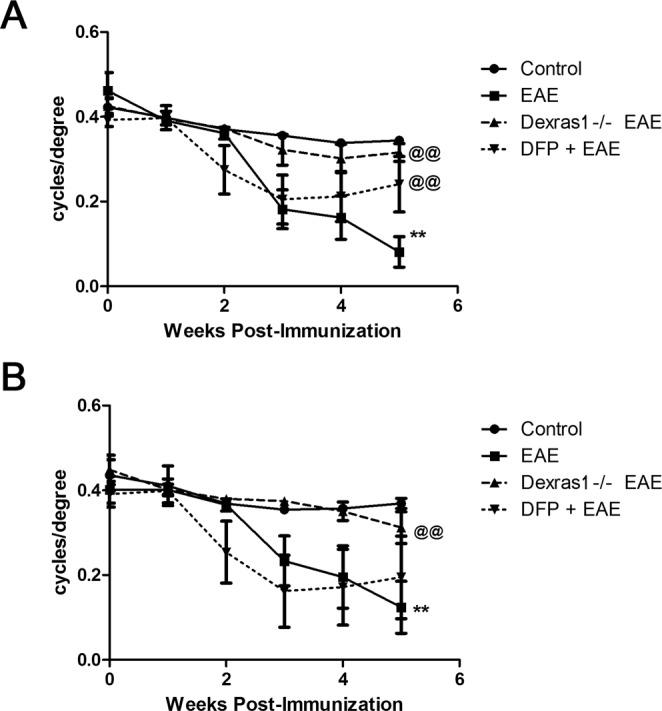
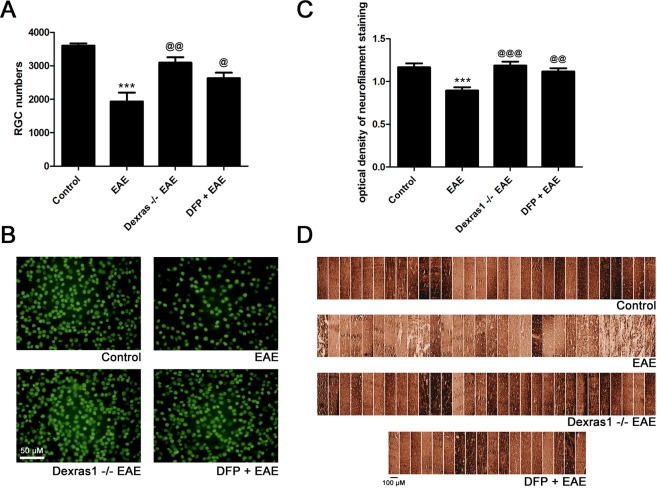
Dysregulation of iron metabolism, and resultant cytotoxicity, has been implicated in the pathogenesis of multiple sclerosis (MS) and other neurodegenerative processes. Iron accumulation promotes cytotoxicity through various mechanisms including oxidative stress and glutamate toxicity, and occurs in both MS patients and in the experimental autoimmune encephalomyelitis (EAE) model of MS. Divalent Metal Transporter1, a major iron importer in cells, is stimulated by signaling of Dexras1, a small G protein member of the Ras family. Dexras1 is activated by S-nitrosylation by nitric oxide (NO) produced by either inducible nitric oxide synthase in activated microglia/macrophages or neuronal nitric oxide synthase in neurons. Here we show Dexras1 exacerbates oxidative stress-induced neurodegeneration in experimental optic neuritis, an inflammatory demyelinating optic nerve condition that occurs in MS and EAE. Dexras1 deletion, as well as treatment with the iron chelator deferiprone, preserves vision and attenuates retinal ganglion cell (RGC) and axonal loss during EAE optic neuritis. These results suggest that iron entry triggered by NO-activated Dexras1 signaling is a potential mechanism of neuronal death in experimental optic neuritis. The current data suggest modulation of Dexras1 signaling and iron chelation are potential novel treatment strategies for optic neuritis and MS, and possibly other optic neuropathies as well.
铁代谢失调,以及由此产生的细胞毒性,与多发性硬化症(MS)和其他神经退行性过程的发病机制有关。铁积累通过多种机制促进细胞毒性,包括氧化应激和谷氨酸毒性,并且在 MS 患者和 MS 的实验性自身免疫性脑脊髓炎(EAE)模型中都发生。二价金属转运蛋白 1(Divalent Metal Transporter 1)是细胞内的主要铁摄取器,受 Ras 家族中小 G 蛋白成员 Dexras1 的信号刺激。Dexras1 通过激活小胶质细胞/巨噬细胞中诱导型一氧化氮合酶或神经元中神经元型一氧化氮合酶产生的一氧化氮(NO)的 S-亚硝基化而被激活。在这里,我们表明 Dexras1 加剧了实验性视神经炎(MS 和 EAE 中发生的炎症性脱髓鞘视神经疾病)中氧化应激诱导的神经退行性变。Dexras1 缺失以及用铁螯合剂地拉罗司(deferiprone)治疗可在 EAE 视神经炎期间保持视力并减轻视网膜神经节细胞(RGC)和轴突丢失。这些结果表明,NO 激活的 Dexras1 信号触发的铁进入是实验性视神经炎中神经元死亡的潜在机制。目前的数据表明,Dexras1 信号转导的调节和铁螯合可能是视神经炎和 MS 以及其他可能的视神经病变的潜在新治疗策略。