Institute of Biotechnology, Life Sciences Center, Vilnius University, Saulėtekio al. 7, Vilnius, 10257, Lithuania.
BMC Bioinformatics. 2019 Aug 13;20(1):419. doi: 10.1186/s12859-019-2913-3.
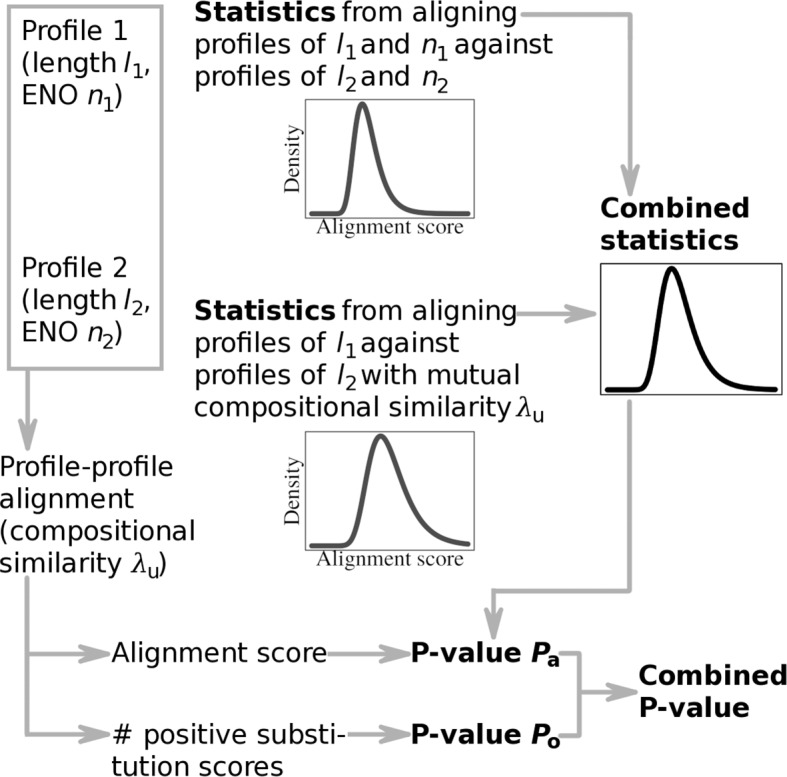
Alignment of sequence families described by profiles provides a sensitive means for establishing homology between proteins and is important in protein evolutionary, structural, and functional studies. In the context of a steadily growing amount of sequence data, estimating the statistical significance of alignments, including profile-profile alignments, plays a key role in alignment-based homology search algorithms. Still, it is an open question as to what and whether one type of distribution governs profile-profile alignment score, especially when profile-profile substitution scores involve such terms as secondary structure predictions.
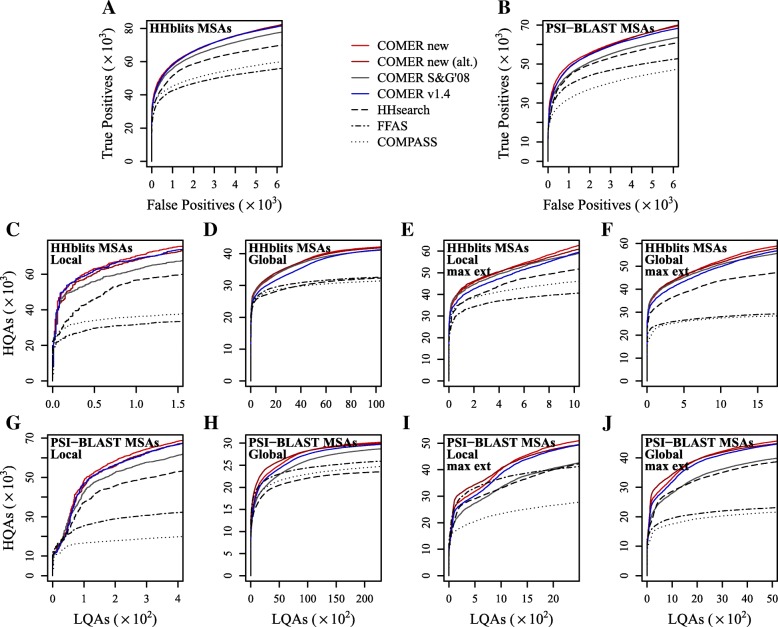
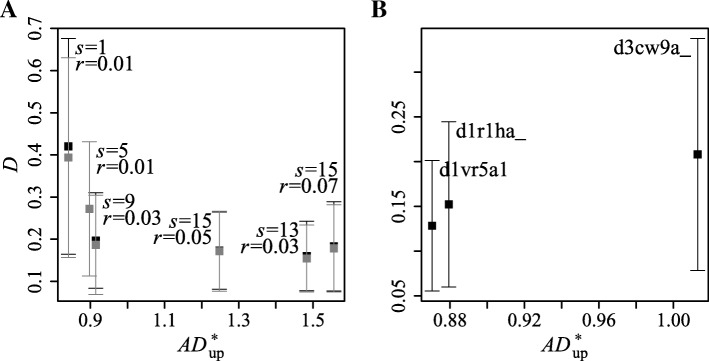
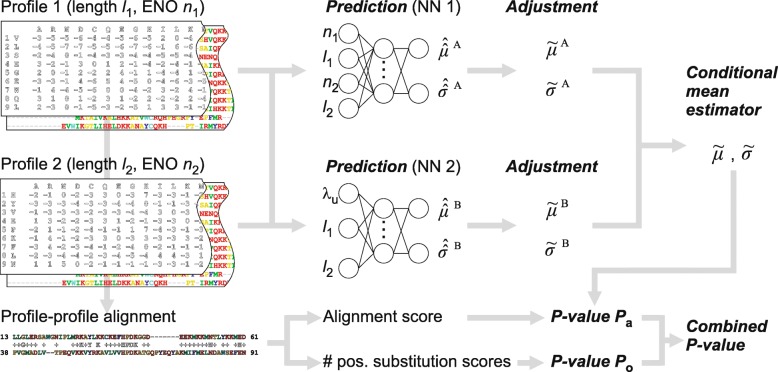
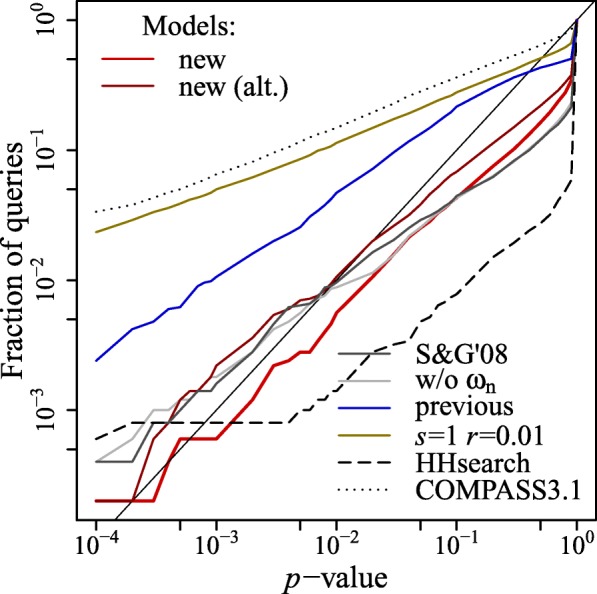
This study presents a methodology for estimating the statistical significance of this type of alignments. The methodology rests on a new algorithm developed for generating random profiles such that their alignment scores are distributed similarly to those obtained for real unrelated profiles. We show that improvements in statistical accuracy and sensitivity and high-quality alignment rate result from statistically characterizing alignments by establishing the dependence of statistical parameters on various measures associated with both individual and pairwise profile characteristics. Implemented in the COMER software, the proposed methodology yielded an increase of up to 34.2% in the number of true positives and up to 61.8% in the number of high-quality alignments with respect to the previous version of the COMER method.
The more accurate estimation of statistical significance is implemented in the COMER method, which is now more sensitive and provides an increased rate of high-quality profile-profile alignments. The results of the present study also suggest directions for future research.
通过轮廓描述的序列家族对齐为蛋白质之间的同源性提供了一种敏感的方法,在蛋白质进化、结构和功能研究中非常重要。在序列数据不断增长的情况下,评估对齐的统计显著性,包括轮廓轮廓对齐,在基于对齐的同源性搜索算法中起着关键作用。然而,什么和是否有一种分布控制轮廓轮廓对齐分数仍然是一个悬而未决的问题,特别是当轮廓轮廓替换分数涉及到二级结构预测等术语时。
本研究提出了一种用于估计这种类型的对齐的统计显著性的方法。该方法基于一种新的算法,用于生成随机轮廓,使得它们的对齐分数与为真实不相关的轮廓获得的分数分布相似。我们表明,通过建立统计参数与与单个和成对轮廓特征相关的各种措施的依赖性,通过统计特征描述对齐来提高统计准确性和敏感性以及高质量对齐率。在 COMER 软件中实现的,所提出的方法与 COMER 方法的前一个版本相比,在真阳性的数量上增加了高达 34.2%,在高质量对齐的数量上增加了高达 61.8%。
COMER 方法实现了更准确的统计显著性估计,现在更敏感,并提供了更高的高质量轮廓轮廓对齐率。本研究的结果还为未来的研究方向提供了建议。