Shteinfer-Kuzmine Anna, Argueti Shirel, Gupta Rajeev, Shvil Neta, Abu-Hamad Salah, Gropper Yael, Hoeber Jan, Magrì Andrea, Messina Angela, Kozlova Elena N, Shoshan-Barmatz Varda, Israelson Adrian
Department of Life Sciences, The National Institute for Biotechnology in the Negev, Ben-Gurion University of the Negev, Beersheba, Israel.
Department of Physiology and Cell Biology, Faculty of Health Sciences, The Zlotowski Center for Neuroscience, Ben-Gurion University of the Negev, Beersheba, Israel.
Front Cell Neurosci. 2019 Aug 14;13:346. doi: 10.3389/fncel.2019.00346. eCollection 2019.
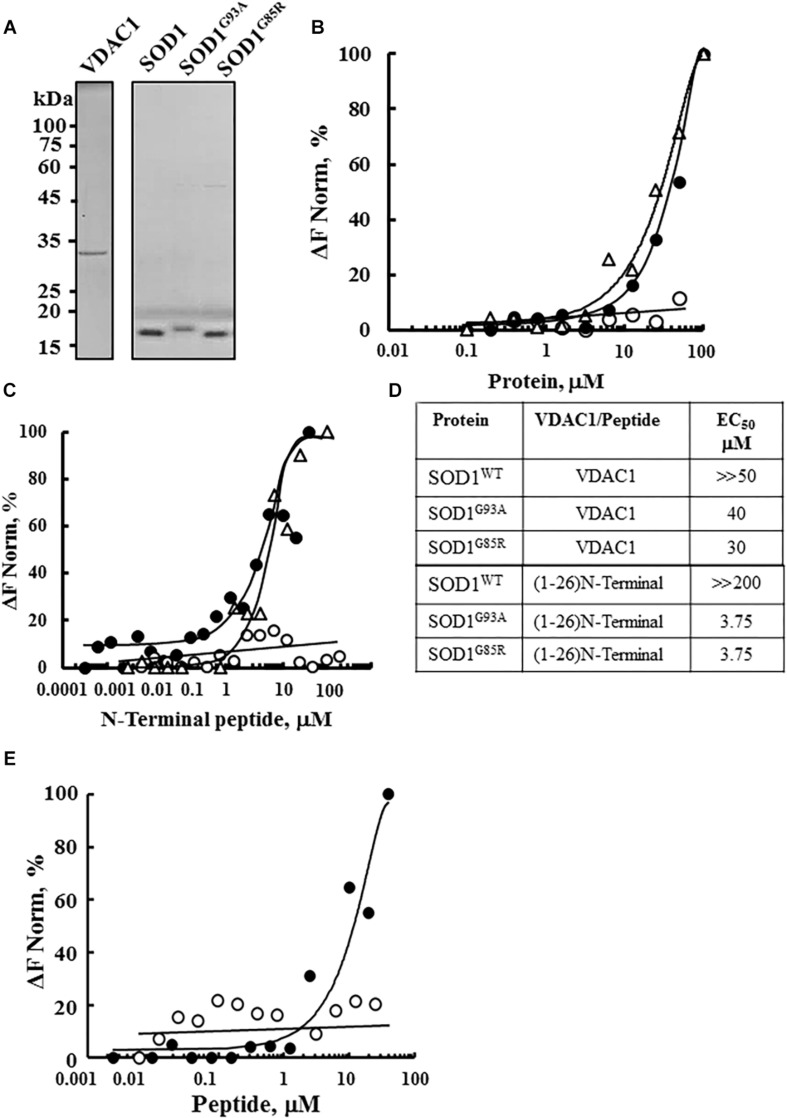
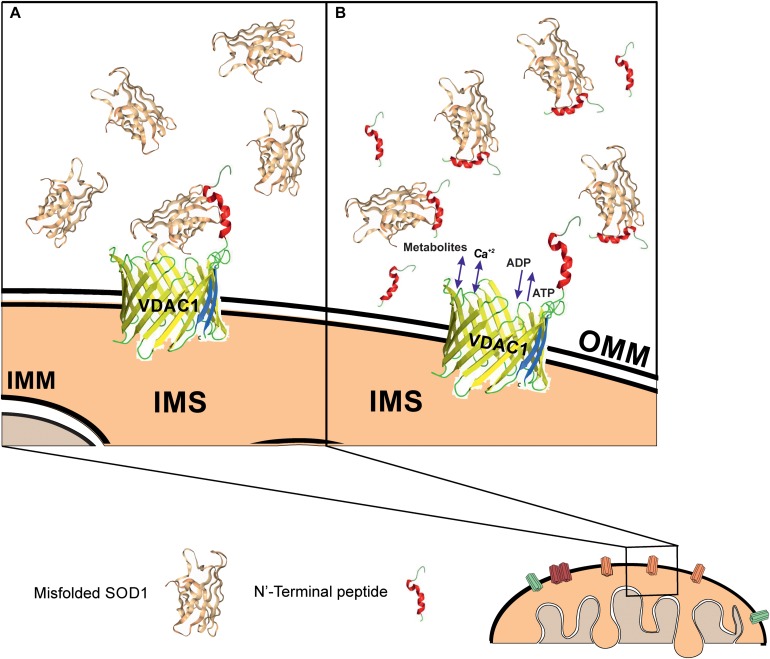
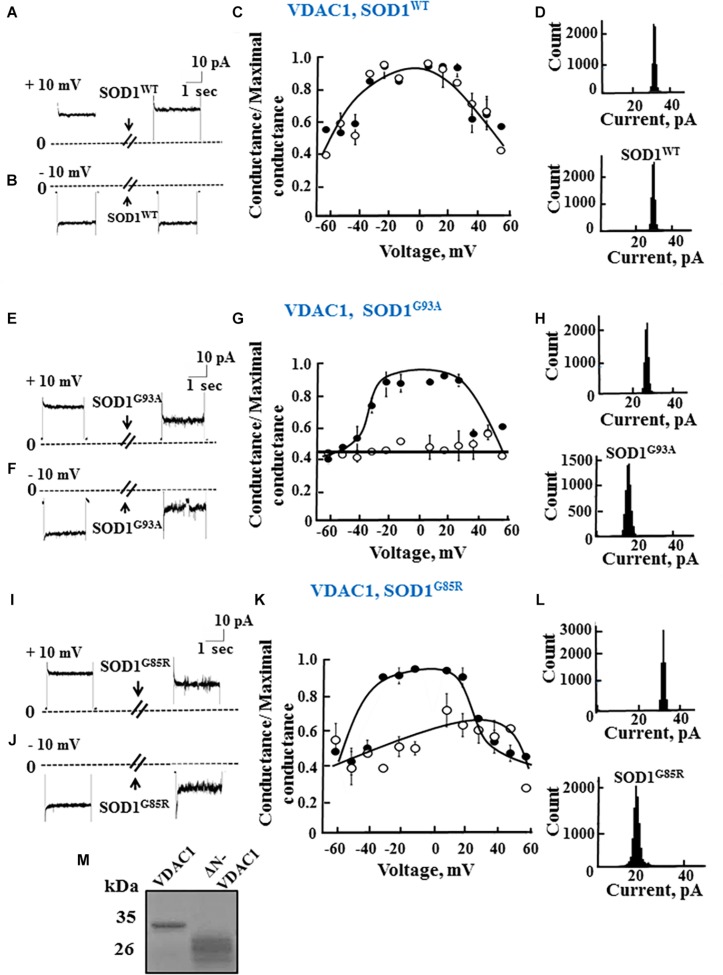
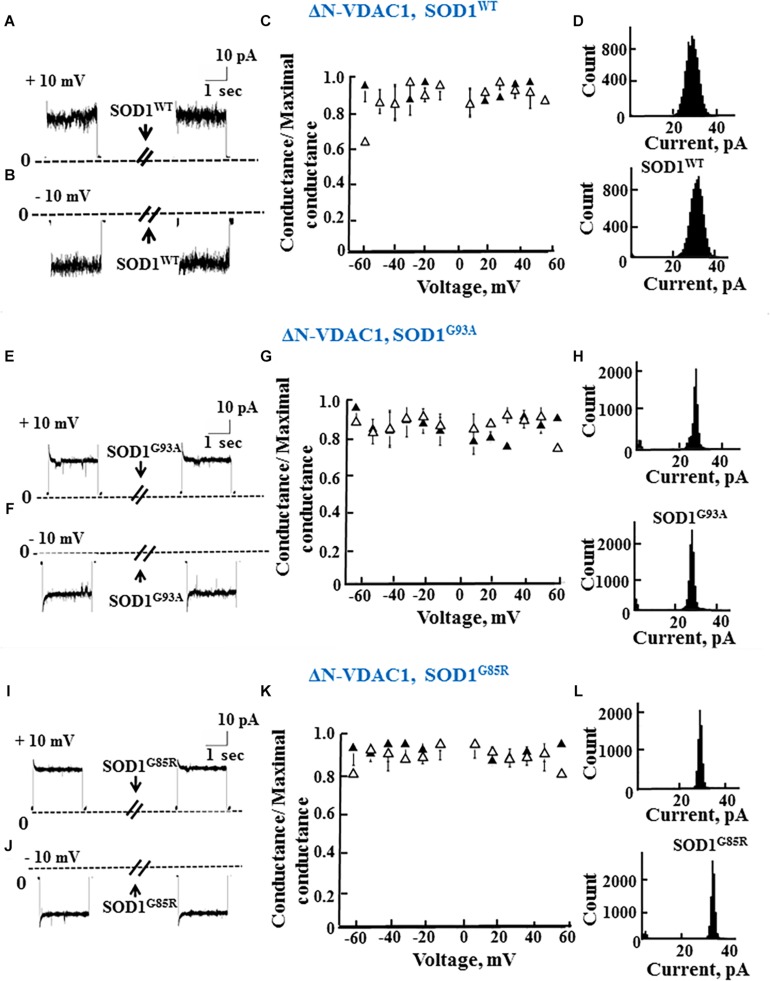
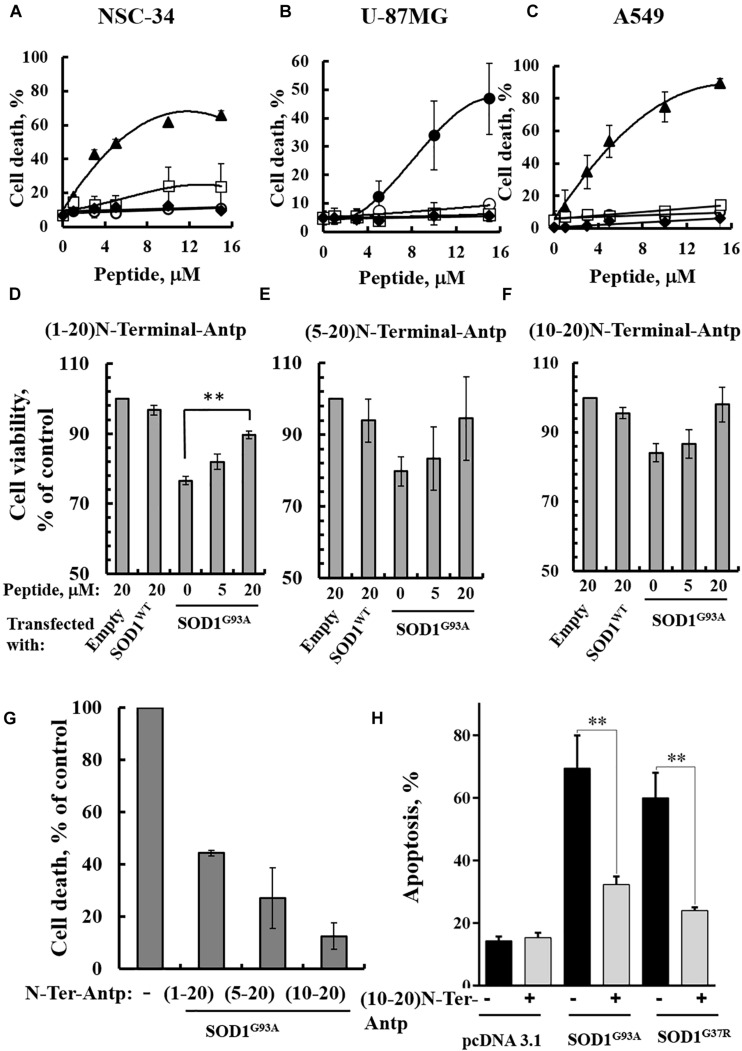
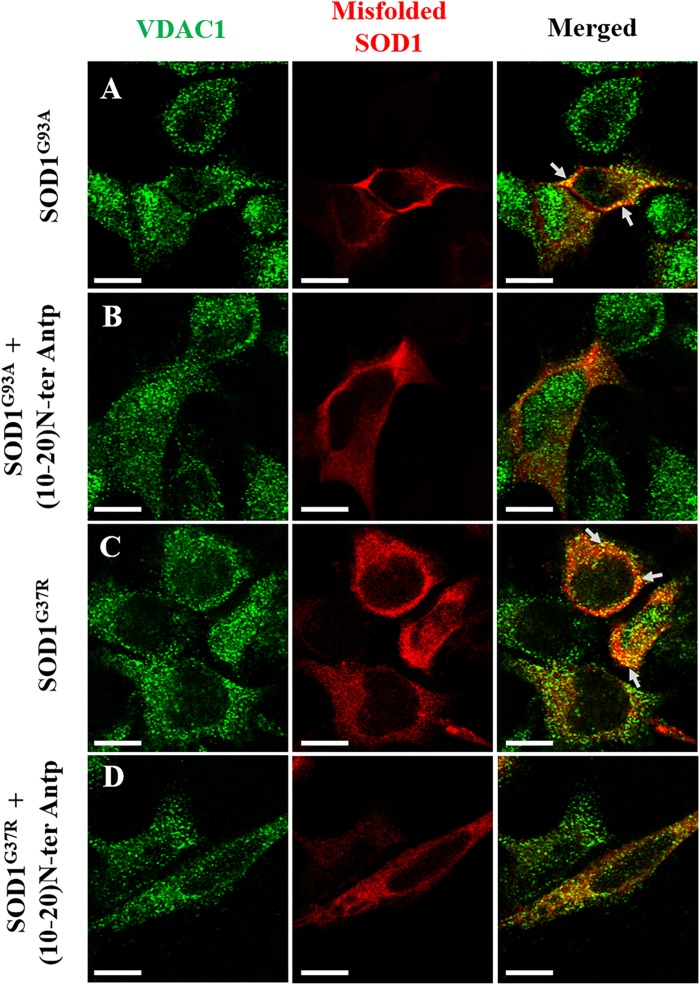
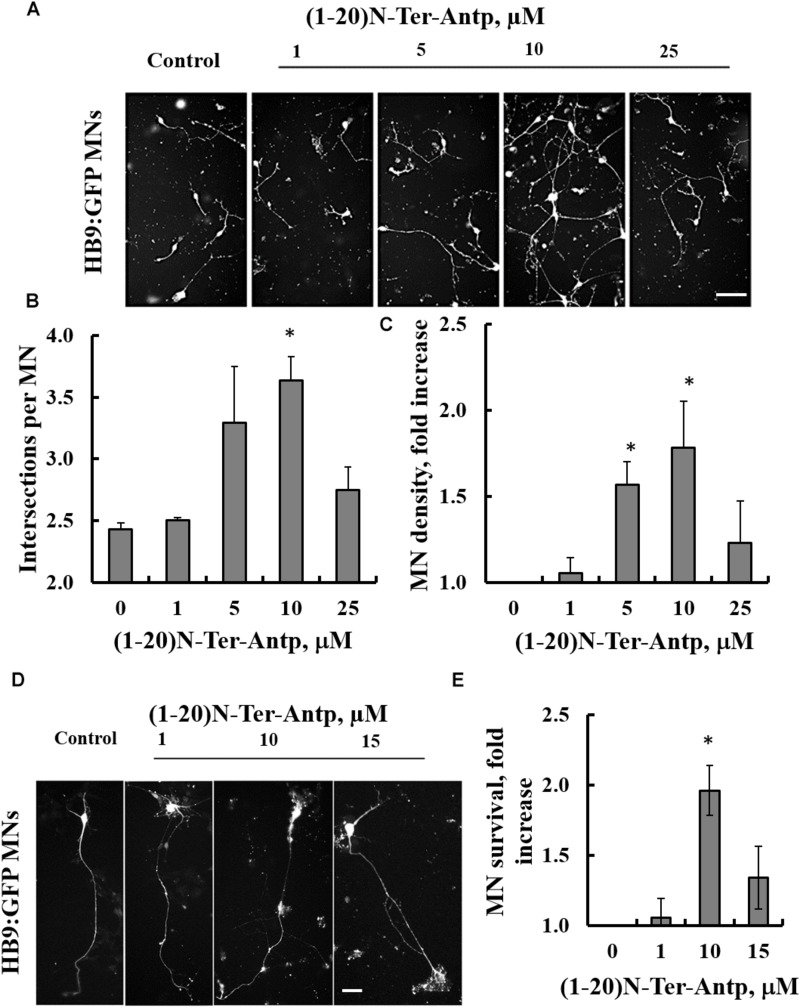
Mutations in superoxide dismutase (SOD1) are the second most common cause of familial amyotrophic lateral sclerosis (ALS), a fatal neurodegenerative disease caused by the death of motor neurons in the brain and spinal cord. SOD1 neurotoxicity has been attributed to aberrant accumulation of misfolded SOD1, which in its soluble form binds to intracellular organelles, such as mitochondria and ER, disrupting their functions. Here, we demonstrate that mutant SOD1 binds specifically to the N-terminal domain of the voltage-dependent anion channel (VDAC1), an outer mitochondrial membrane protein controlling cell energy, metabolic and survival pathways. Mutant SOD1 and SOD1, but not wild type SOD1, directly interact with VDAC1 and reduce its channel conductance. No such interaction with N-terminal-truncated VDAC1 occurs. Moreover, a VDAC1-derived N-terminal peptide inhibited mutant SOD1-induced toxicity. Incubation of motor neuron-like NSC-34 cells expressing mutant SOD1 or mouse embryonic stem cell-derived motor neurons with different VDAC1 N-terminal peptides resulted in enhanced cell survival. Taken together, our results establish a direct link between mutant SOD1 toxicity and the VDAC1 N-terminal domain and suggest that VDAC1 N-terminal peptides targeting mutant SOD1 provide potential new therapeutic strategies for ALS.
超氧化物歧化酶(SOD1)突变是家族性肌萎缩侧索硬化症(ALS)的第二大常见病因,ALS是一种由大脑和脊髓中运动神经元死亡导致的致命性神经退行性疾病。SOD1神经毒性被认为是由于错误折叠的SOD1异常积累所致,其可溶形式与细胞内细胞器(如线粒体和内质网)结合,破坏其功能。在此,我们证明突变型SOD1特异性结合电压依赖性阴离子通道(VDAC1)的N端结构域,VDAC1是一种控制细胞能量、代谢和生存途径的线粒体外膜蛋白。突变型SOD1和SOD1(而非野生型SOD1)直接与VDAC1相互作用并降低其通道电导。与N端截短的VDAC1不存在这种相互作用。此外,一种源自VDAC1的N端肽可抑制突变型SOD1诱导的毒性。用不同的VDAC1 N端肽孵育表达突变型SOD1的运动神经元样NSC-34细胞或小鼠胚胎干细胞衍生的运动神经元,可提高细胞存活率。综上所述,我们的结果建立了突变型SOD1毒性与VDAC1 N端结构域之间的直接联系,并表明靶向突变型SOD1的VDAC1 N端肽为ALS提供了潜在的新治疗策略。