Molina Pilar, Sanz-Sánchez Jorge, Fenollosa Manuel, Martínez-Matilla Marina, Giner Juan, Zorio Esther
Servicio de Patología, Instituto de Medicina Legal y Ciencias Forenses de Valencia, Valencia, España.
Unidad de Valoración del Riesgo de Muerte Súbita Familiar and Unidad de Cardiopatías Familiares, Muerte Súbita y Mecanismos de Enfermedad (CaFaMuSMe), Instituto de Investigación Sanitaria La Fe, Valencia, España.
Forensic Sci Res. 2019 Aug 19;4(3):274-279. doi: 10.1080/20961790.2019.1616247. eCollection 2019.
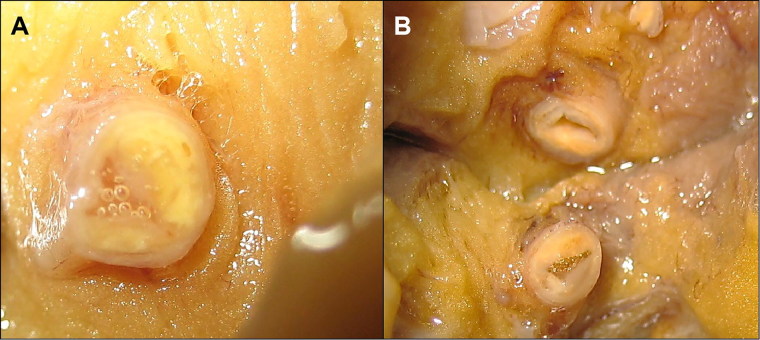
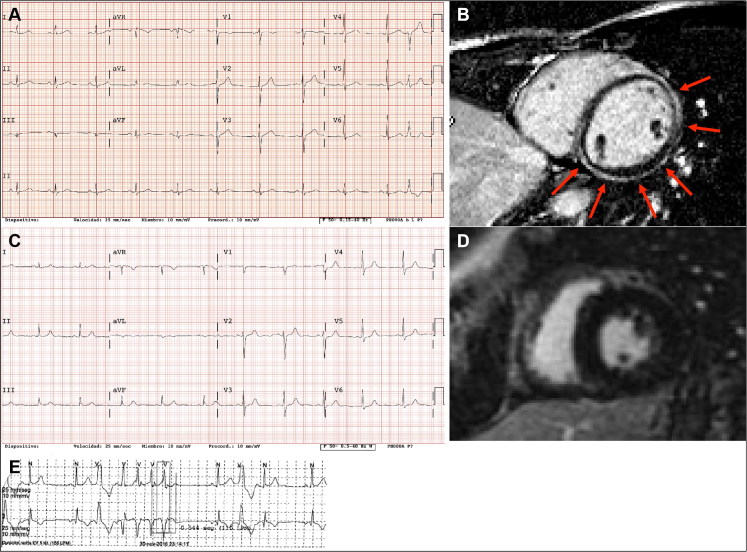
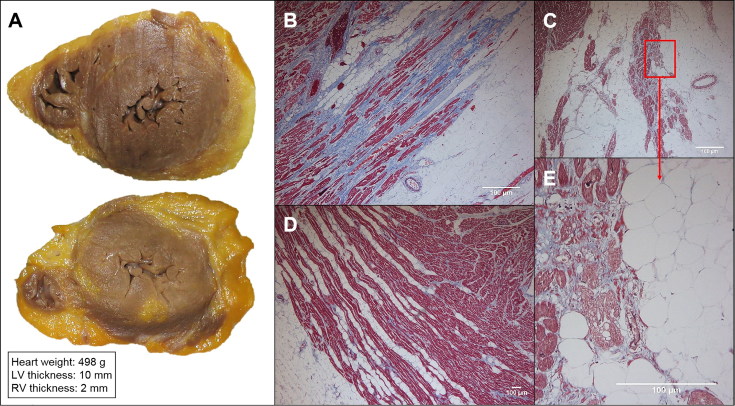
Ischemic heart disease (IHD) is the leading cause of sudden cardiac death (SCD) and often non-thrombosed severe coronary stenoses with or without myocardial scars are detected. Left dominant arrhythmogenic cardiomyopathy (LDAC) is a life-threating rare disease which has been more thoroughly studied in the last 10 years. The macroscopic study of an SCD victim was conducted and re-evaluated 9 years later. The cardiological work-up in his first-degree relatives initially comprised an electrocardiogram (ECG) and an echocardiogram. When they were re-evaluted 9 years later, a cardiac magnetic resonance, an ECG-monitoring, an exercise testing and a genetic study were performed and the pedigree was extended accordingly. In 2008, an IHD was suspected in the sports-triggered SCD of a 37-year-old man upon the postmortem (75% stenosis of the left main and circumflex coronary arteries; the subepicardial left ventricular fibrofatty infiltration with mild myocardial degeneration was assumed to be a past myocardial infarction). No cardiomyopathy was identified in any of the two proband's sisters. Nine years thereafter, distant relatives were diagnosed with LDAC due to a pathogenic desmoplakin mutation. The reanalysis of the two sisters showed ventricular arrhythmias in one of them without structural heart involvement and the reviewed postmortem of the proband was reclassified as LDAC based on the fibrofatty infiltration; both were mutation carriers. The completion of the family study on 19 family members yielded one SCD due to LDAC (the proband), three living patients diagnosed with LDAC (two with a defibrillator), one mutation carrier without structural ventricular involvement, and 14 healthy relatives (who were discharged) with a very good co-segregation of the mutation. Although rare, LDAC exists and sometimes its differential diagnosis with IHD has to be faced. Modifying previous postmortem misdiagnoses can help family screening to further prevent SCDs.
缺血性心脏病(IHD)是心源性猝死(SCD)的主要原因,常可检测到无血栓形成的严重冠状动脉狭窄,伴或不伴有心肌瘢痕。左优势型致心律失常性心肌病(LDAC)是一种危及生命的罕见疾病,在过去10年中得到了更深入的研究。对一名SCD受害者进行了大体研究,并在9年后进行了重新评估。其一级亲属最初的心脏检查包括心电图(ECG)和超声心动图。9年后对他们进行重新评估时,进行了心脏磁共振成像、心电图监测、运动试验和基因研究,并相应地扩展了家系。2008年,一名37岁男性在运动诱发的SCD后尸检时怀疑患有IHD(左主干和回旋支冠状动脉狭窄75%;心外膜下左心室纤维脂肪浸润伴轻度心肌变性被认为是既往心肌梗死)。两名先证者的姐妹均未发现心肌病。9年后,远亲因致病性桥粒斑蛋白突变被诊断为LDAC。对两姐妹的重新分析显示,其中一人有室性心律失常,但无结构性心脏受累,根据纤维脂肪浸润,先证者经复查的尸检结果被重新分类为LDAC;两人均为突变携带者。对19名家庭成员的家系研究完成后发现,有1例因LDAC导致的SCD(先证者),3例确诊为LDAC的在世患者(2例植入了除颤器),1例无结构性心室受累的突变携带者,以及14名健康亲属(已出院),该突变的共分离情况良好。尽管LDAC罕见,但它确实存在,有时必须面对其与IHD的鉴别诊断。修正先前的尸检误诊有助于家族筛查,以进一步预防SCD。