Guffon Nathalie, Froissart Roseline, Fouilhoux Alain
Centre de Référence des Maladies Héréditaires du Métabolisme, Hôpital Femme Mère Enfant Hospices Civils de Lyon Bron France.
JIMD Rep. 2019 Jul 29;49(1):1-6. doi: 10.1002/jmd2.12039. eCollection 2019 Sep.
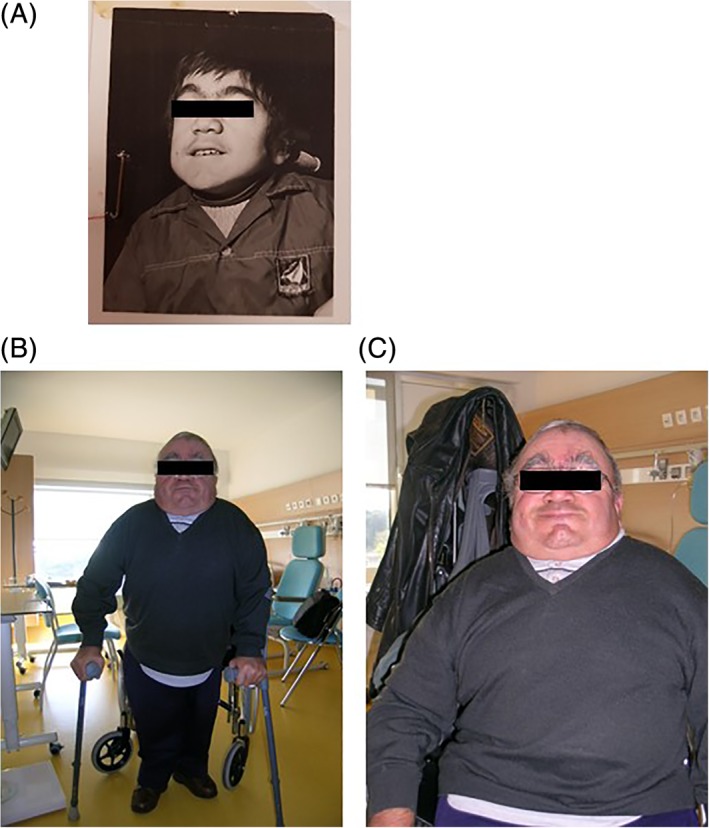
Mucopolysaccharidoses VII, or Sly syndrome, is linked to mutations in the beta-glucuronidase encoding gene. Sly syndrome is a rare condition and presentation is highly variable, ranging from a prenatal form with severe, lethal fetal hydrops to more benign adolescent or adult forms with simple thoracic kyphosis. Molecular diagnosis of this adult male patient identified two missense mutations in the gene that led to a deficiency in beta-glucuronidase catalytic activity and the resulting accumulation of chondroitin sulfate glycosaminoglycans. During childhood, bilateral inguinal hernia was repaired at 1 year of age and gait abnormalities were noted, leading to a bilateral femoral varization osteotomy due to a bilateral coxa valga with hip subluxation at the age of 7.5. The patient suffered regular upper respiratory infections and required numerous orthopedic surgeries. Despite learning difficulties with visual and hearing deficits, the patient worked full-time and undertook regular leisure activities. At 33 years of age, the patient's health deteriorated; a hip replacement and glaucoma leading to reductions in his visual field limited his capacity to travel independently. The patient was hospitalized at 51. Although he remained self-sufficient for taking meals, he needed help with many daily activities. Following a period marked by major asthenia with a general loss of autonomy, the patient died at 52 years of age. With the advent of new enzyme replacement therapies, this medical history of this rare untreated attenuated patient may provide benchmarks to judge the efficacy of treatment in future patients.
黏多糖贮积症VII型,即斯利综合征,与β-葡萄糖醛酸酶编码基因突变有关。斯利综合征是一种罕见疾病,临床表现高度多变,从伴有严重致死性胎儿水肿的产前型到伴有单纯胸椎后凸的较为良性的青少年或成人型。对这名成年男性患者的分子诊断发现该基因存在两个错义突变,导致β-葡萄糖醛酸酶催化活性缺乏,进而引起硫酸软骨素糖胺聚糖蓄积。患儿1岁时接受双侧腹股沟疝修补术,当时发现步态异常,7.5岁时因双侧髋外翻伴髋关节半脱位接受双侧股骨内翻截骨术。患者经常患上部呼吸道感染,需要多次接受骨科手术。尽管存在视觉和听力缺陷导致学习困难,但患者仍全职工作并定期参加休闲活动。33岁时,患者健康状况恶化;髋关节置换术和青光眼导致视野缩小,限制了他独立出行的能力。51岁时患者住院。尽管他仍能自行用餐,但许多日常活动需要帮助。在经历了一段以极度虚弱和普遍自主能力丧失为特征的时期后,患者于52岁去世。随着新的酶替代疗法的出现,这名未经治疗的罕见轻症患者的病史可能为判断未来患者的治疗效果提供基准。