Centre of New Technologies, Warsaw 02-097, Poland.
Department of Mathematics, Informatics and Mechanics, University of Warsaw, Warsaw 02-097, Poland.
Bioinformatics. 2020 Feb 1;36(3):953-955. doi: 10.1093/bioinformatics/btz644.
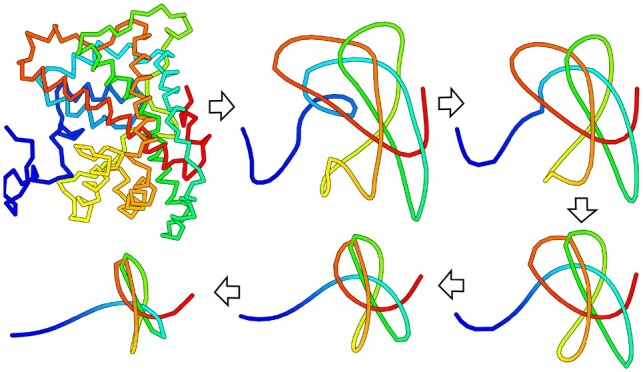
The biggest hurdle in studying topology in biopolymers is the steep learning curve for actually seeing the knots in structure visualization. Knot_pull is a command line utility designed to simplify this process-it presents the user with a smoothing trajectory for provided structures (any number and length of protein, RNA or chromatin chains in PDB, CIF or XYZ format), and calculates the knot type (including presence of any links, and slipknots when a subchain is specified).
Knot_pull works under Python >=2.7 and is system independent. Source code and documentation are available at http://github.com/dzarmola/knot_pull under GNU GPL license and include also a wrapper script for PyMOL for easier visualization. Examples of smoothing trajectories can be found at: https://www.youtube.com/watch?v=IzSGDfc1vAY.
Supplementary data are available at Bioinformatics online.
在生物聚合物中研究拓扑结构的最大障碍是实际观察结构可视化中的结时陡峭的学习曲线。Knot_pull 是一个命令行实用程序,旨在简化此过程 - 它为用户提供了一个平滑的轨迹,用于提供的结构(任何数量和长度的蛋白质,RNA 或染色质链,以 PDB,CIF 或 XYZ 格式),并计算结类型(包括任何链接的存在,以及指定子链时的滑结)。
Knot_pull 在 Python >=2.7 下运行,与系统无关。源代码和文档可在 http://github.com/dzarmola/knot_pull 下获得,根据 GNU GPL 许可证可用,并且还包括用于 PyMOL 的包装脚本,以便于可视化。平滑轨迹的示例可在:https://www.youtube.com/watch?v=IzSGDfc1vAY 找到。
补充数据可在 Bioinformatics 在线获得。