Institute for Translation Research in Biomedicine, Kaohsiung Chang Gung Memorial Hospital, Kaohsiung, Taiwan.
Research Center for Neuroscience, Institute of Molecular Biosciences, Mahidol University, Nakhon Pathom, Thailand.
Neuropathol Appl Neurobiol. 2020 Jun;46(4):375-390. doi: 10.1111/nan.12584. Epub 2019 Nov 6.
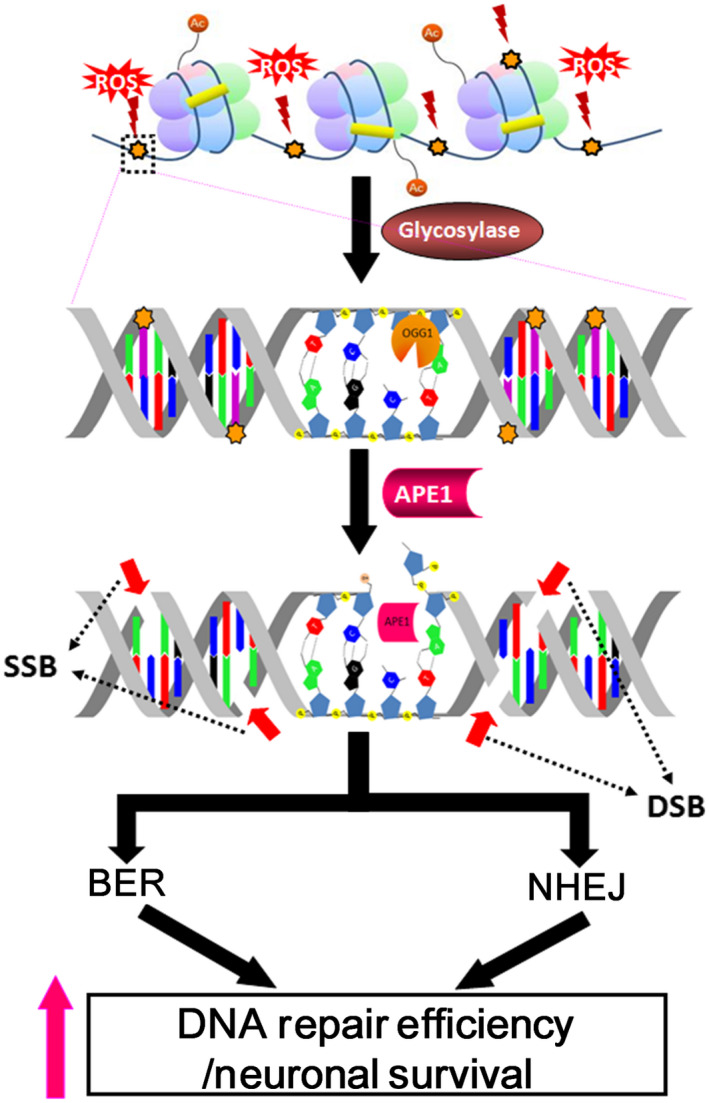
Accumulating studies have suggested that base excision repair (BER) is the major repair pathway of oxidative DNA damage in neurons, and neurons are deficient in other DNA repair pathways, including nucleotide excision repair and homologous recombination repair. However, some studies have demonstrated that neurons could efficiently repair glutamate- and menadione-induced double-strand breaks (DSBs), suggesting that the DSB repair mechanisms might be implicated in neuronal health. In this study, we hypothesized that BER and nonhomologous end joining (NHEJ) work together to repair oxidative DNA damage in neurons.
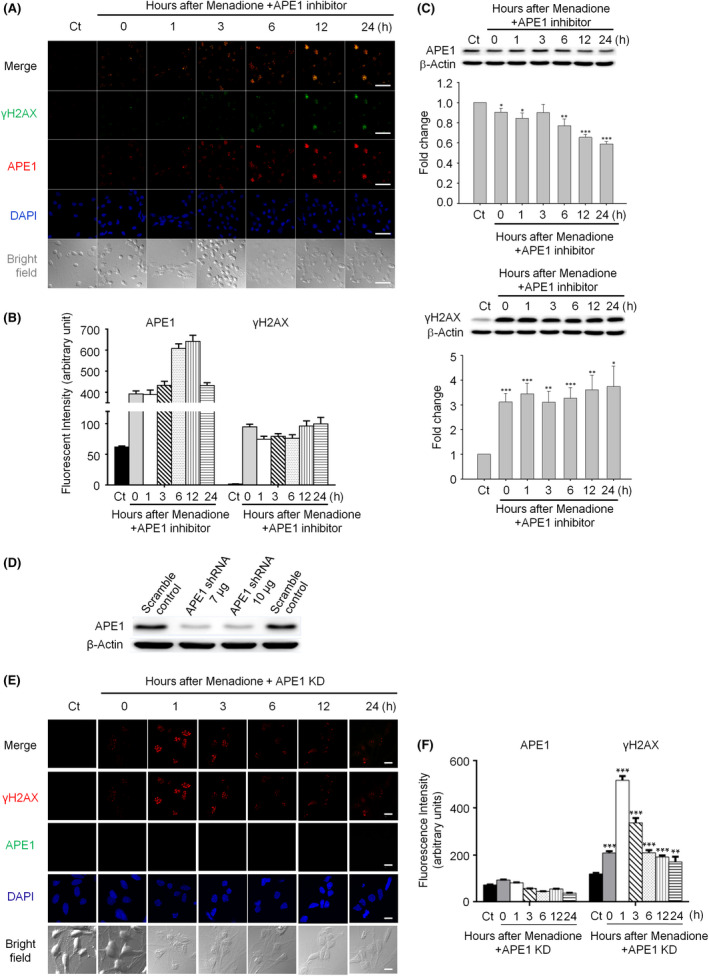
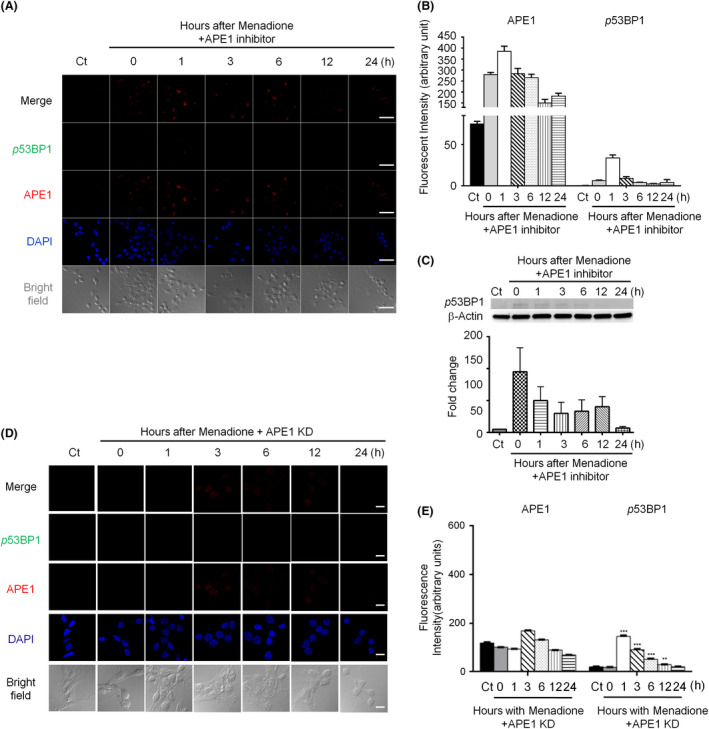
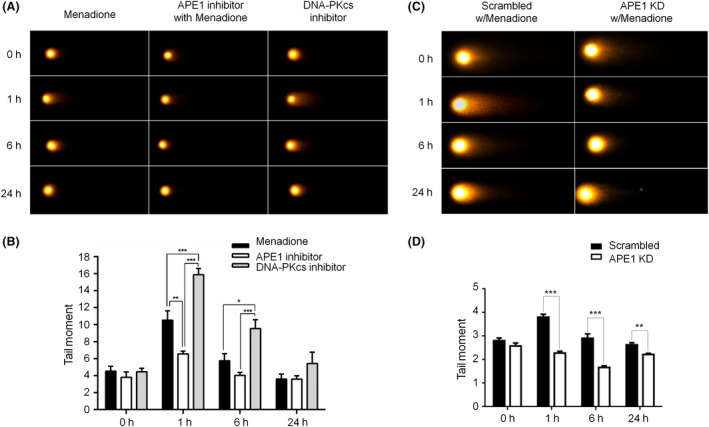
Immunohistochemistry and confocal microscopy were employed to examine the colocalization of apyrimidinic endonuclease 1 (APE1), histone variant 2AX (γH2AX) and phosphorylated p53-binding protein (53BP1). APE1 inhibitor and shRNA were respectively applied to suppress APE1 activity and protein expression to determine the correlation of APE1 and DSB formation. The neutral comet assay was used to determine and quantitate the formation of DSB.
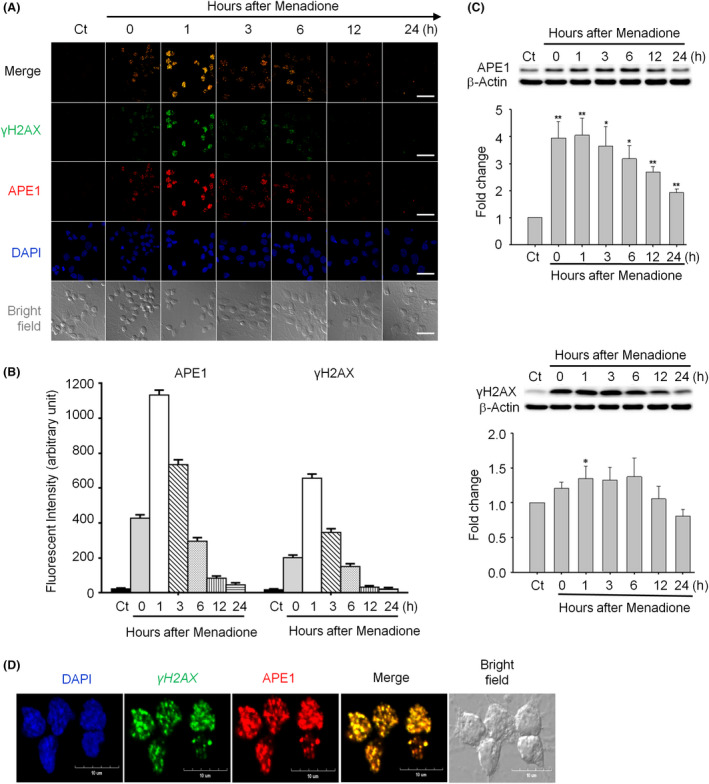
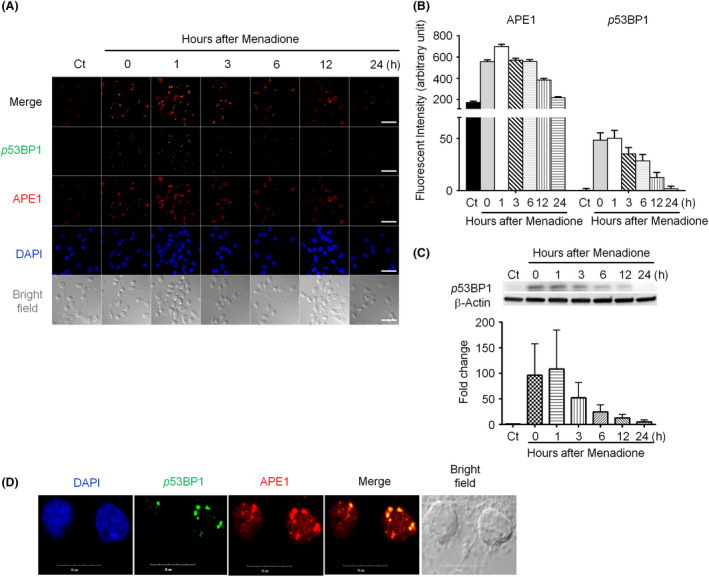
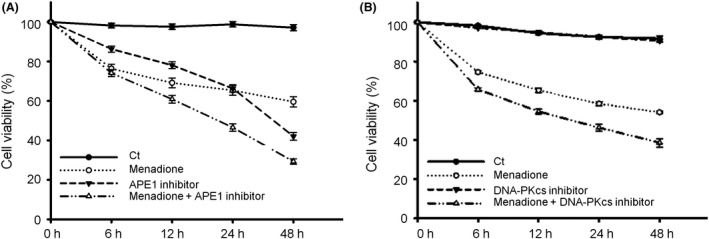
Both γH2AX and 53BP1 were upregulated and colocalized with APE1 in the nuclei of rat cortical neurons subjected to menadione-induced oxidative insults. Phospho53BP1 foci were efficiently abolished, but γH2AX foci persisted following the suppression of APE1 activity. Comet assays demonstrated that the inhibition of APE1 decreased the DSB formation.
Our results indicate that APE1 can engage the NHEJ mechanism in the repair of oxidative DNA damage in neurons. These findings provide insights into the mechanisms underlying the efficient repair of oxidative DNA damage in neurons despite the high oxidative burden.
越来越多的研究表明,碱基切除修复(BER)是神经元氧化 DNA 损伤的主要修复途径,而神经元缺乏其他 DNA 修复途径,包括核苷酸切除修复和同源重组修复。然而,一些研究表明,神经元可以有效地修复谷氨酸和亚甲蓝诱导的双链断裂(DSBs),这表明 DSB 修复机制可能与神经元健康有关。在这项研究中,我们假设 BER 和非同源末端连接(NHEJ)共同修复神经元中的氧化 DNA 损伤。
免疫组织化学和共聚焦显微镜用于检测脱嘌呤/脱嘧啶内切酶 1(APE1)、组蛋白变体 2AX(γH2AX)和磷酸化 p53 结合蛋白(53BP1)的共定位。分别应用 APE1 抑制剂和 shRNA 抑制 APE1 活性和蛋白表达,以确定 APE1 与 DSB 形成的相关性。中性彗星试验用于确定和定量 DSB 的形成。
亚甲蓝诱导的氧化应激后,大鼠皮质神经元细胞核中 γH2AX 和 53BP1 均上调,并与 APE1 共定位。磷酸化 53BP1 焦点被有效消除,但 APE1 活性抑制后 γH2AX 焦点仍然存在。彗星试验表明,APE1 的抑制减少了 DSB 的形成。
我们的结果表明,APE1 可以在神经元氧化 DNA 损伤的修复中参与 NHEJ 机制。这些发现为尽管氧化应激高,但神经元中氧化 DNA 损伤有效修复的机制提供了深入的了解。