College of Information Science & Technology, University of Nebraska at Omaha, Omaha, Nebraska, United States of America.
Department of Biochemistry and Molecular Biology, University of Nebraska Medical Center, Omaha, Nebraska, United States of America.
PLoS Comput Biol. 2019 Oct 25;15(10):e1007469. doi: 10.1371/journal.pcbi.1007469. eCollection 2019 Oct.
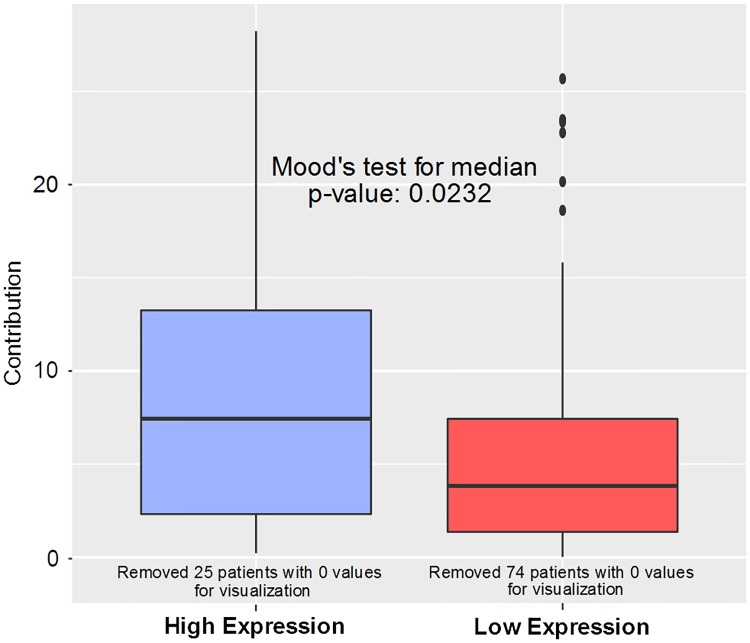
Splice variants have been shown to play an important role in tumor initiation and progression and can serve as novel cancer biomarkers. However, the clinical importance of individual splice variants and the mechanisms by which they can perturb cellular functions are still poorly understood. To address these issues, we developed an efficient and robust computational method to: (1) identify splice variants that are associated with patient survival in a statistically significant manner; and (2) predict rewired protein-protein interactions that may result from altered patterns of expression of such variants. We applied our method to the lung adenocarcinoma dataset from TCGA and identified splice variants that are significantly associated with patient survival and can alter protein-protein interactions. Among these variants, several are implicated in DNA repair through homologous recombination. To computationally validate our findings, we characterized the mutational signatures in patients, grouped by low and high expression of a splice variant associated with patient survival and involved in DNA repair. The results of the mutational signature analysis are in agreement with the molecular mechanism suggested by our method. To the best of our knowledge, this is the first attempt to build a computational approach to systematically identify splice variants associated with patient survival that can also generate experimentally testable, mechanistic hypotheses. Code for identifying survival-significant splice variants using the Null Empirically Estimated P-value method can be found at https://github.com/thecodingdoc/neep. Code for construction of Multi-Granularity Graphs to discover potential rewired protein interactions can be found at https://github.com/scwest/SINBAD.
剪接变异体已被证明在肿瘤的发生和发展中起着重要作用,并可以作为新的癌症生物标志物。然而,单个剪接变异体的临床意义以及它们如何干扰细胞功能的机制仍知之甚少。为了解决这些问题,我们开发了一种高效、稳健的计算方法来:(1)以统计学上显著的方式识别与患者生存相关的剪接变异体;(2)预测可能由这些变异体表达模式改变引起的重排的蛋白质-蛋白质相互作用。我们将我们的方法应用于 TCGA 的肺腺癌数据集,并确定了与患者生存显著相关且可改变蛋白质-蛋白质相互作用的剪接变异体。在这些变异体中,有几个通过同源重组参与 DNA 修复。为了对我们的发现进行计算机验证,我们对患者进行了特征分析,根据与患者生存相关且涉及 DNA 修复的剪接变异体的低表达和高表达进行分组。突变特征分析的结果与我们方法所建议的分子机制一致。据我们所知,这是首次尝试构建一种计算方法来系统地识别与患者生存相关的剪接变异体,这些变异体也可以产生可在实验中检验的、基于机制的假说。使用 Null Empirically Estimated P-value 方法识别生存显著剪接变异体的代码可在 https://github.com/thecodingdoc/neep 上找到。用于构建多粒度图以发现潜在重排蛋白质相互作用的代码可在 https://github.com/scwest/SINBAD 上找到。