Key Laboratory of Textile Science & Technology of Ministry of Education, College of Textiles, Donghua University, Shanghai 201620, China.
Shanghai Naturalism Biological Technology Co., Ltd., Shanghai 201616, China.
Molecules. 2019 Oct 24;24(21):3831. doi: 10.3390/molecules24213831.
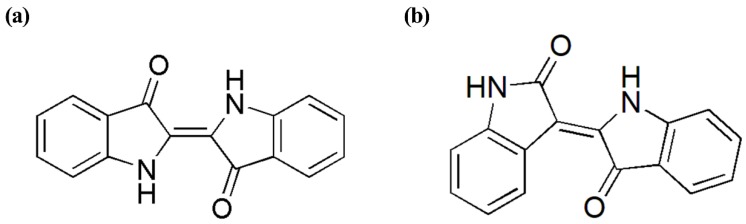
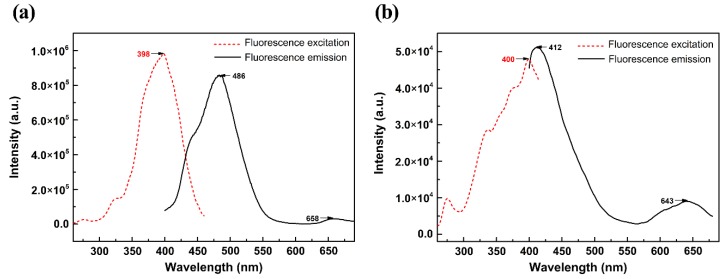
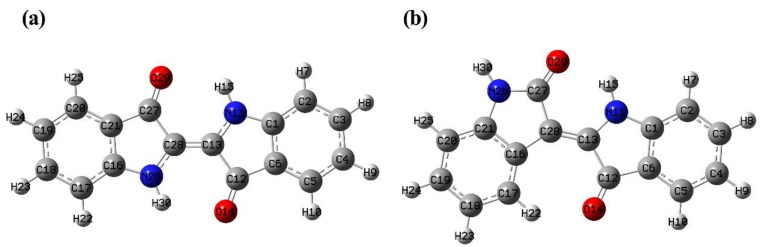
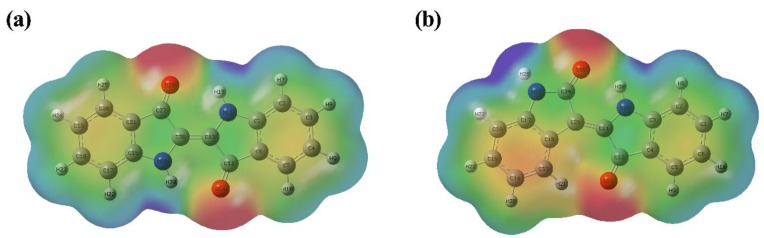
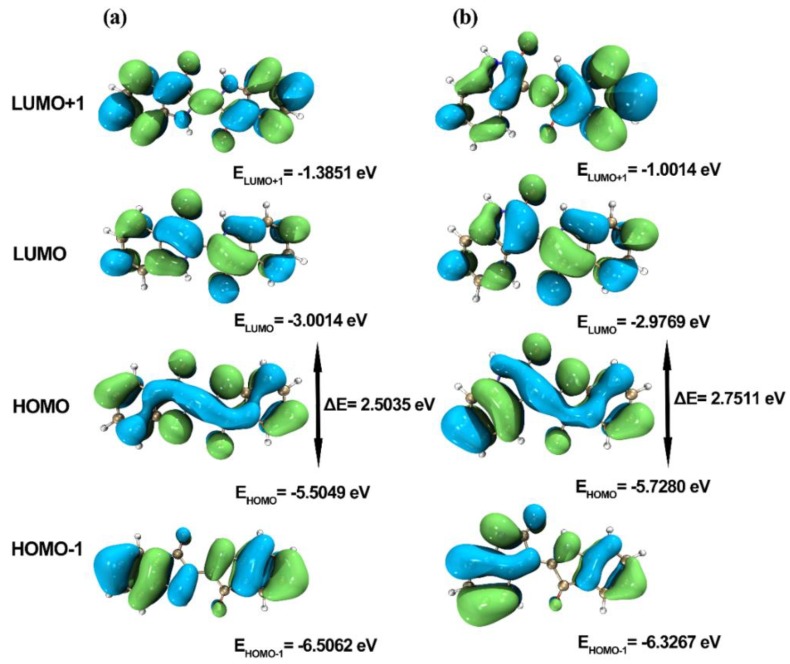
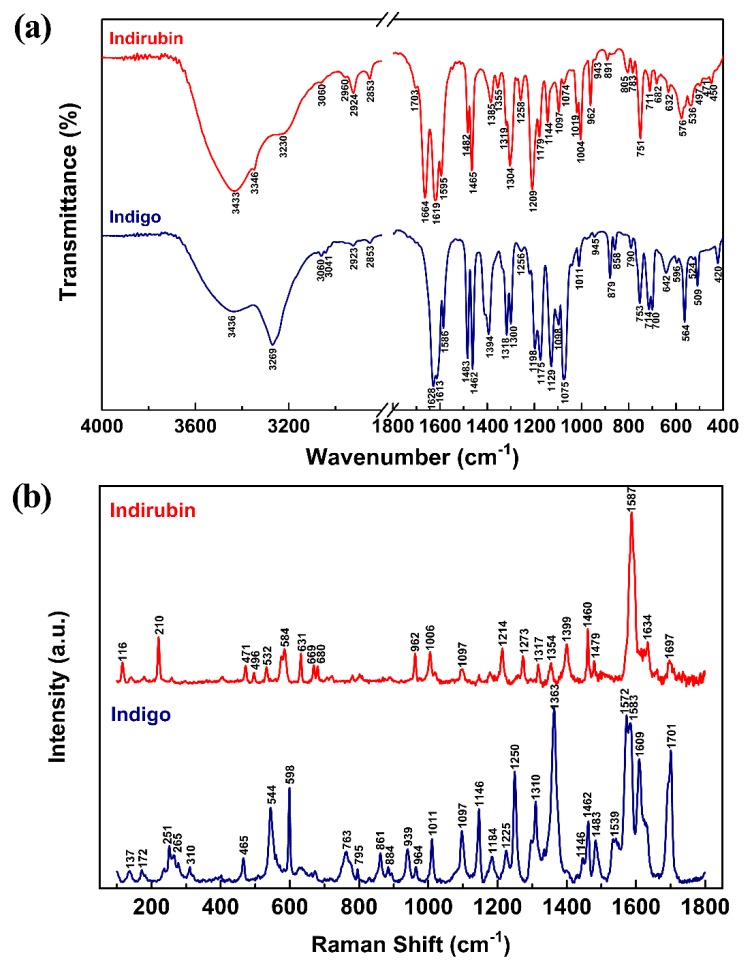
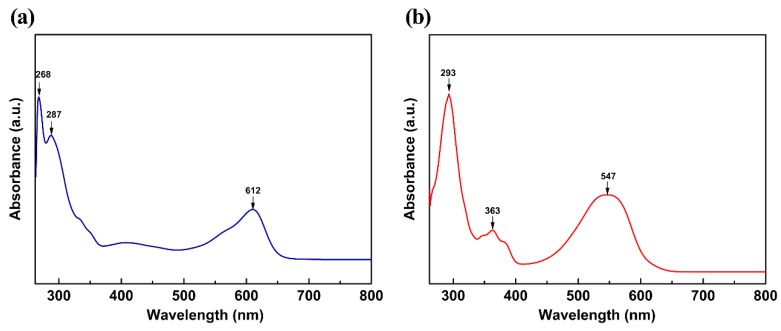
This paper presents a comparative study on natural indigo and indirubin in terms of molecular structures and spectral properties by using both computational and experimental methods. The spectral properties were analyzed with Fourier transform infrared (FTIR), Raman, UV-Visible, and fluorescence techniques. The density functional theory (DFT) method with B3LYP using 6-311G(d,p) basis set was utilized to obtain their optimized geometric structures and calculate the molecular electrostatic potential, frontier molecular orbitals, FTIR, and Raman spectra. The single-excitation configuration interaction (CIS), time-dependent density functional theory (TD-DFT), and polarization continuum model (PCM) were used to optimize the excited state structure and calculate the UV-Visible absorption and fluorescence spectra of the two molecules at B3LYP/6-311G(d,p) level. The results showed that all computational spectra agreed well with the experimental results. It was found that the same vibrational mode presents a lower frequency in indigo than that in indirubin. The frontier molecular orbital analysis demonstrated that the UV-Visible absorption and fluorescence bands of indigo and indirubin are mainly derived from π → π* transition. The results also implied that the indigo molecule is more conjugated and planar than indirubin, thereby exhibiting a longer maximum absorption wavelength and stronger fluorescence peak.
本文通过计算和实验方法,对天然靛蓝和靛玉红在分子结构和光谱性质方面进行了比较研究。利用傅里叶变换红外(FTIR)、拉曼、紫外-可见和荧光技术分析了光谱性质。采用 B3LYP 方法和 6-311G(d,p)基组,对其优化的几何结构进行了计算,并计算了分子静电势、前线分子轨道、FTIR 和拉曼光谱。采用单激发组态相互作用(CIS)、含时密度泛函理论(TD-DFT)和极化连续模型(PCM),在 B3LYP/6-311G(d,p)水平上对激发态结构进行了优化,并计算了两种分子的紫外-可见吸收和荧光光谱。结果表明,所有计算光谱都与实验结果吻合良好。发现靛蓝中相同的振动模式的频率低于靛玉红。前线分子轨道分析表明,靛蓝和靛玉红的紫外-可见吸收和荧光带主要来源于π→π*跃迁。结果还表明,靛蓝分子比靛玉红更共轭和平面化,因此表现出更长的最大吸收波长和更强的荧光峰。