Department of Chemistry and Biology, Ryerson University, 350 Victoria St, Toronto, M5B 2K3, Canada.
SciNet High Performance Computing Consortium, University of Toronto, 661 University Ave, Toronto, M5G 1M1, Canada.
BMC Bioinformatics. 2019 Oct 29;20(1):533. doi: 10.1186/s12859-019-3100-2.
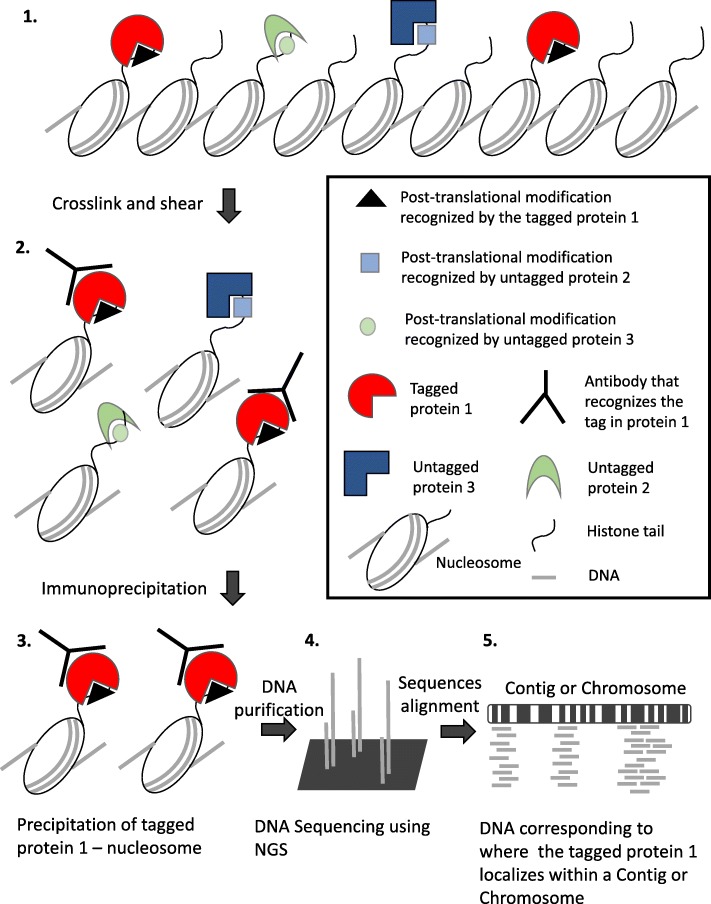
Chromatin immunoprecipitation coupled to next generation sequencing (ChIP-Seq) is a widely-used molecular method to investigate the function of chromatin-related proteins by identifying their associated DNA sequences on a genomic scale. ChIP-Seq generates large quantities of data that is difficult to process and analyze, particularly for organisms with a contig-based sequenced genomes that typically have minimal annotation on their associated set of genes other than their associated coordinates primarily predicted by gene finding programs. Poorly annotated genome sequence makes comprehensive analysis of ChIP-Seq data difficult and as such standardized analysis pipelines are lacking.
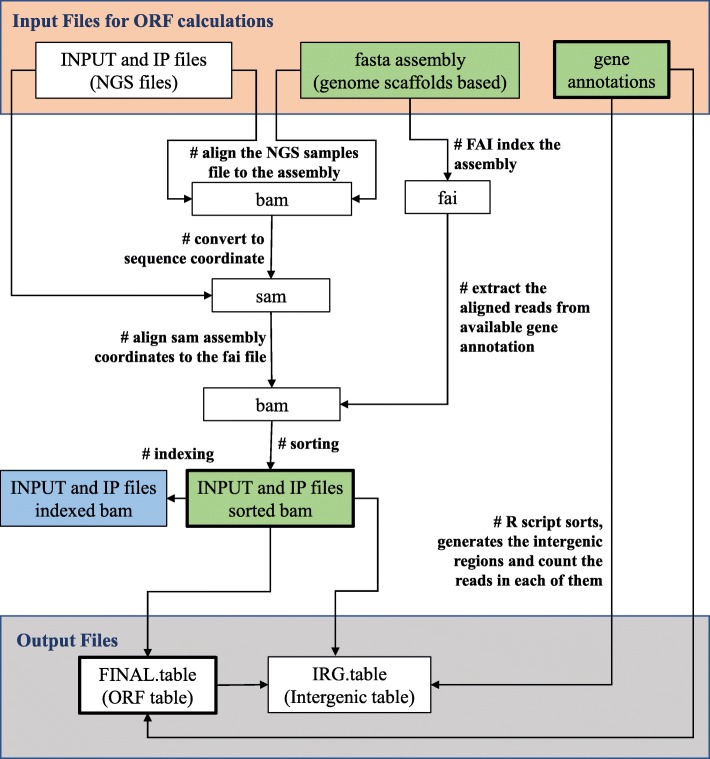
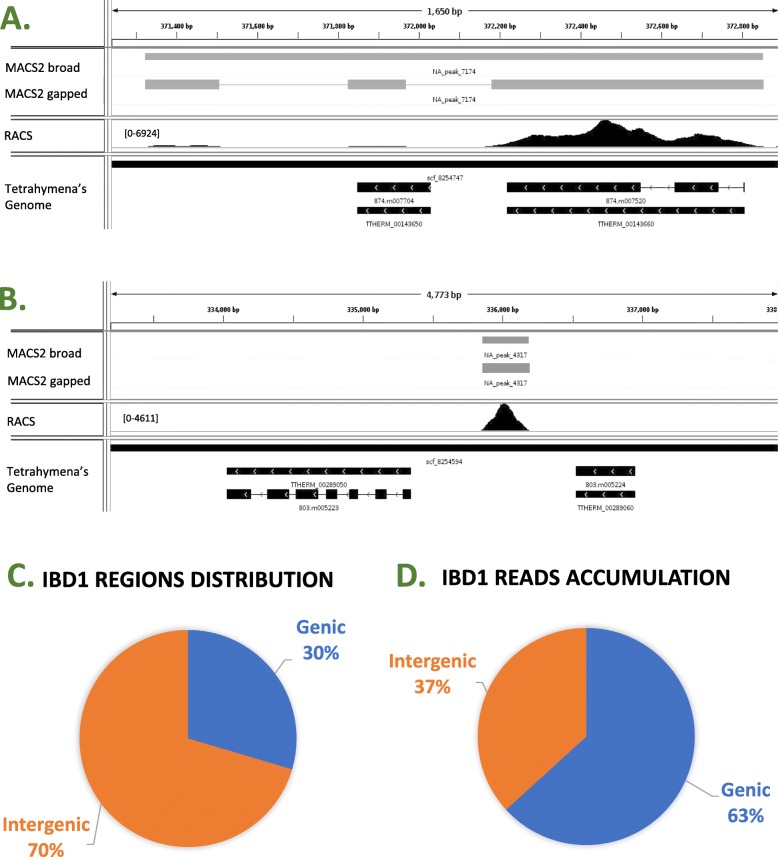
We present a one-stop computational pipeline, "Rapid Analysis of ChIP-Seq data" (RACS), that utilizes traditional High-Performance Computing (HPC) techniques in association with open source tools for processing and analyzing raw ChIP-Seq data. RACS is an open source computational pipeline available from any of the following repositories https://bitbucket.org/mjponce/RACS or https://gitrepos.scinet.utoronto.ca/public/?a=summary&p=RACS . RACS is particularly useful for ChIP-Seq in organisms with contig-based genomes that have poor gene annotation to aid protein function discovery.To test the performance and efficiency of RACS, we analyzed ChIP-Seq data previously published in a model organism Tetrahymena thermophila which has a contig-based genome. We assessed the generality of RACS by analyzing a previously published data set generated using the model organism Oxytricha trifallax, whose genome sequence is also contig-based with poor annotation.
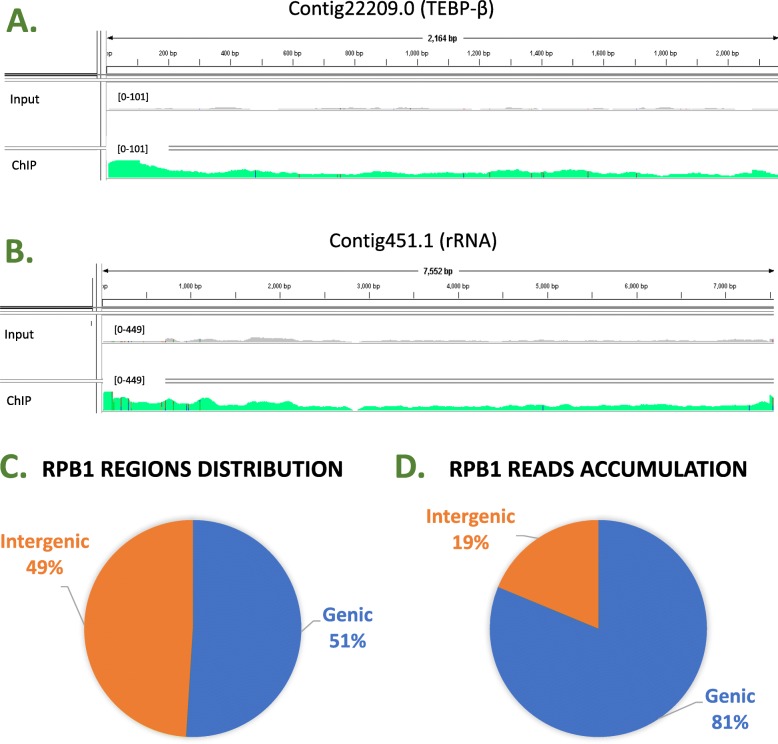
The RACS computational pipeline presented in this report is an efficient and reliable tool to analyze genome-wide raw ChIP-Seq data generated in model organisms with poorly annotated contig-based genome sequence. Because RACS segregates the found read accumulations between genic and intergenic regions, it is particularly efficient for rapid downstream analyses of proteins involved in gene expression.
染色质免疫沉淀结合下一代测序(ChIP-Seq)是一种广泛使用的分子方法,通过在基因组范围内鉴定染色质相关蛋白的相关 DNA 序列来研究染色质相关蛋白的功能。ChIP-Seq 会生成大量的数据,这些数据很难处理和分析,尤其是对于基于连续体的测序基因组的生物体,这些生物体除了主要由基因发现程序预测的相关坐标之外,很少对其相关基因集进行注释。较差的基因组序列注释使得对 ChIP-Seq 数据的全面分析变得困难,因此缺乏标准化的分析流程。
我们提出了一个一站式计算流程“Rapid Analysis of ChIP-Seq data”(RACS),该流程利用传统的高性能计算(HPC)技术,结合开源工具来处理和分析原始 ChIP-Seq 数据。RACS 是一个开源计算流程,可从以下任何一个存储库中获得:https://bitbucket.org/mjponce/RACS 或 https://gitrepos.scinet.utoronto.ca/public/?a=summary&p=RACS。RACS 特别适用于基于连续体的基因组的生物体的 ChIP-Seq,这些生物体的基因注释较差,有助于蛋白质功能的发现。为了测试 RACS 的性能和效率,我们分析了之前在 Tetrahymena thermophila 模型生物中发表的 ChIP-Seq 数据,该生物具有基于连续体的基因组。我们通过分析之前使用基于连续体的基因组和较差注释的模型生物 Oxytricha trifallax 生成的已发表数据集来评估 RACS 的通用性。
本报告中提出的 RACS 计算流程是一种高效可靠的工具,可用于分析基于连续体的基因组序列注释较差的模型生物中产生的全基因组原始 ChIP-Seq 数据。由于 RACS 将发现的读取积累在基因和非基因区域之间进行划分,因此它特别适合快速进行涉及基因表达的蛋白质的下游分析。