Division of Biomedical Statistics and Informatics, Department of Health Sciences Research, Mayo Clinic, 200 1st St SW, Rochester, MN 55905, USA.
BMC Bioinformatics. 2014 Aug 15;15(1):280. doi: 10.1186/1471-2105-15-280.
Chromatin immunoprecipitation (ChIP) followed by next-generation sequencing (ChIP-Seq) has been widely used to identify genomic loci of transcription factor (TF) binding and histone modifications. ChIP-Seq data analysis involves multiple steps from read mapping and peak calling to data integration and interpretation. It remains challenging and time-consuming to process large amounts of ChIP-Seq data derived from different antibodies or experimental designs using the same approach. To address this challenge, there is a need for a comprehensive analysis pipeline with flexible settings to accelerate the utilization of this powerful technology in epigenetics research.
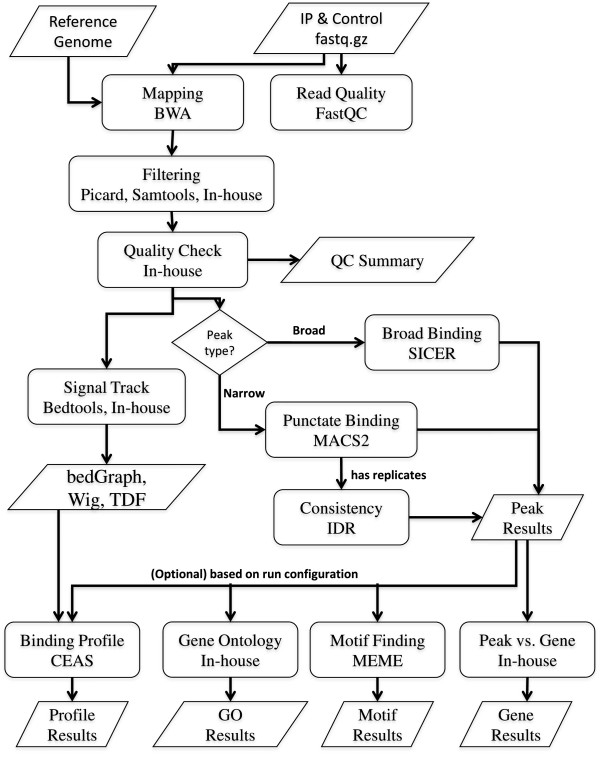
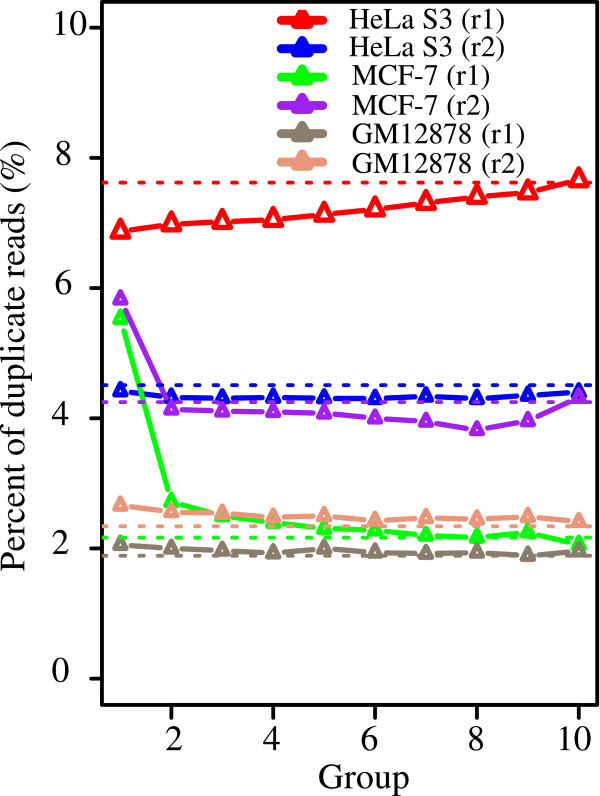
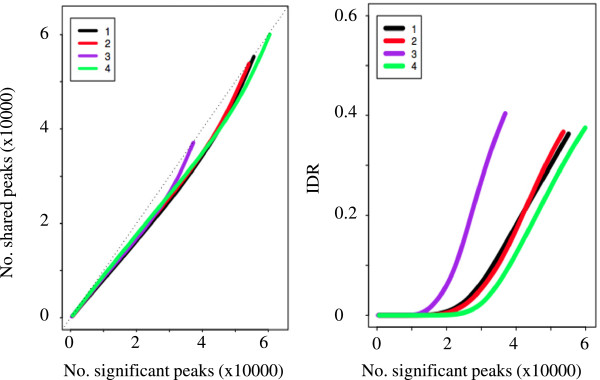
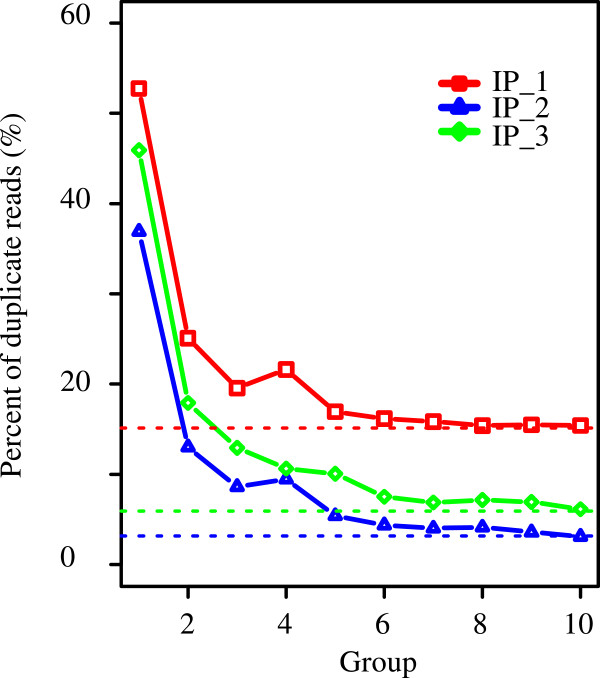
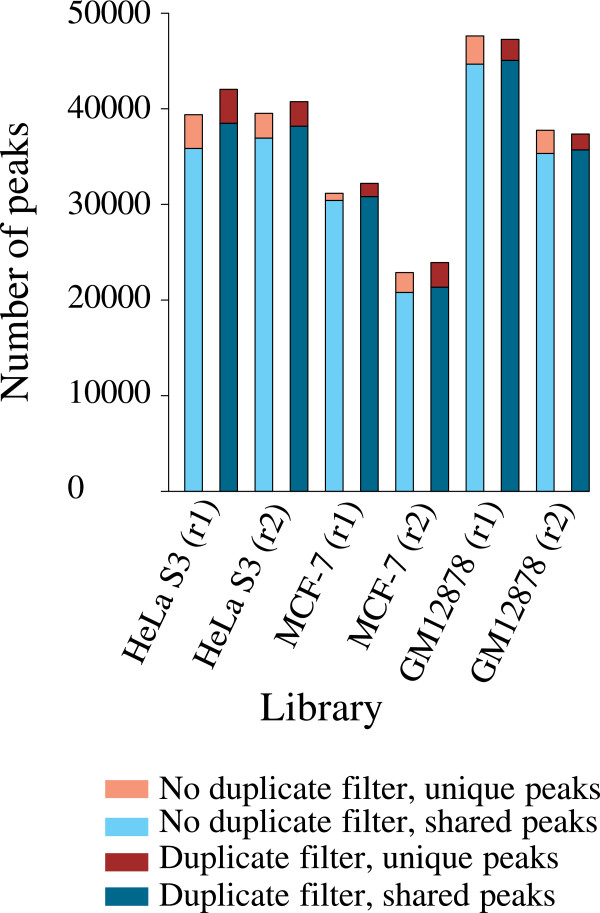
We have developed a highly integrative pipeline, termed HiChIP for systematic analysis of ChIP-Seq data. HiChIP incorporates several open source software packages selected based on internal assessments and published comparisons. It also includes a set of tools developed in-house. This workflow enables the analysis of both paired-end and single-end ChIP-Seq reads, with or without replicates for the characterization and annotation of both punctate and diffuse binding sites. The main functionality of HiChIP includes: (a) read quality checking; (b) read mapping and filtering; (c) peak calling and peak consistency analysis; and (d) result visualization. In addition, this pipeline contains modules for generating binding profiles over selected genomic features, de novo motif finding from transcription factor (TF) binding sites and functional annotation of peak associated genes.
HiChIP is a comprehensive analysis pipeline that can be configured to analyze ChIP-Seq data derived from varying antibodies and experiment designs. Using public ChIP-Seq data we demonstrate that HiChIP is a fast and reliable pipeline for processing large amounts of ChIP-Seq data.
染色质免疫沉淀(ChIP)结合下一代测序(ChIP-Seq)已广泛用于鉴定转录因子(TF)结合和组蛋白修饰的基因组位点。ChIP-Seq 数据分析涉及多个步骤,从读取映射和峰调用到数据集成和解释。使用相同的方法处理来自不同抗体或实验设计的大量 ChIP-Seq 数据仍然具有挑战性和耗时。为了解决这个挑战,需要一个具有灵活设置的综合分析管道,以加速该强大技术在表观遗传学研究中的应用。
我们开发了一个高度综合的管道,称为 HiChIP,用于系统分析 ChIP-Seq 数据。HiChIP 结合了几个基于内部评估和已发表比较选择的开源软件包。它还包括一组内部开发的工具。该工作流程能够分析具有或不具有重复的配对末端和单末端 ChIP-Seq 读取,用于点状和弥散结合位点的特征和注释。HiChIP 的主要功能包括:(a)读取质量检查;(b)读取映射和过滤;(c)峰调用和峰一致性分析;(d)结果可视化。此外,该管道还包含用于在选定的基因组特征上生成绑定配置文件、从转录因子(TF)结合位点发现从头基序和峰相关基因的功能注释的模块。
HiChIP 是一个全面的分析管道,可以配置为分析来自不同抗体和实验设计的 ChIP-Seq 数据。使用公共 ChIP-Seq 数据,我们证明 HiChIP 是一种快速可靠的管道,可处理大量的 ChIP-Seq 数据。