The Jackson Laboratory for Genomic Medicine, Farmington, CT, 06032, USA.
Ohio State University Comprehensive Cancer Center, Columbus, OH, 43210, USA.
Nat Commun. 2019 Nov 6;10(1):5029. doi: 10.1038/s41467-019-13036-1.
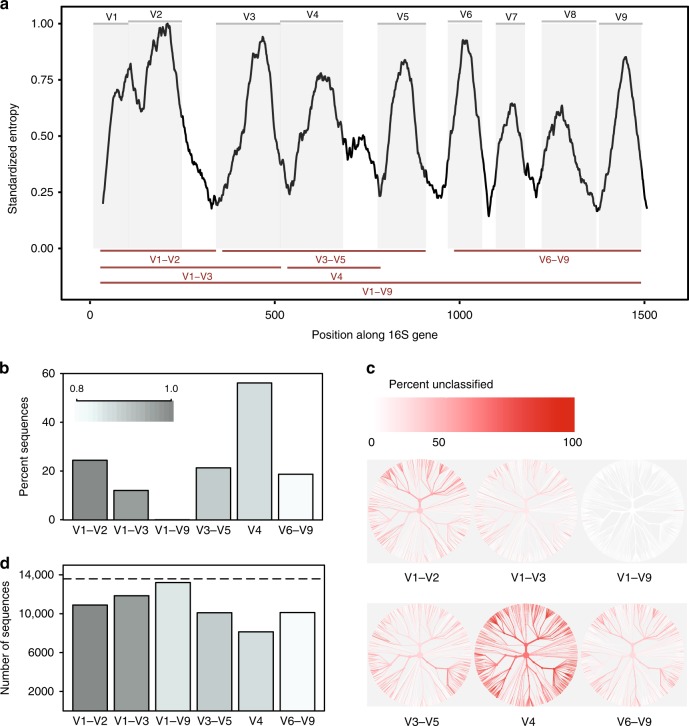
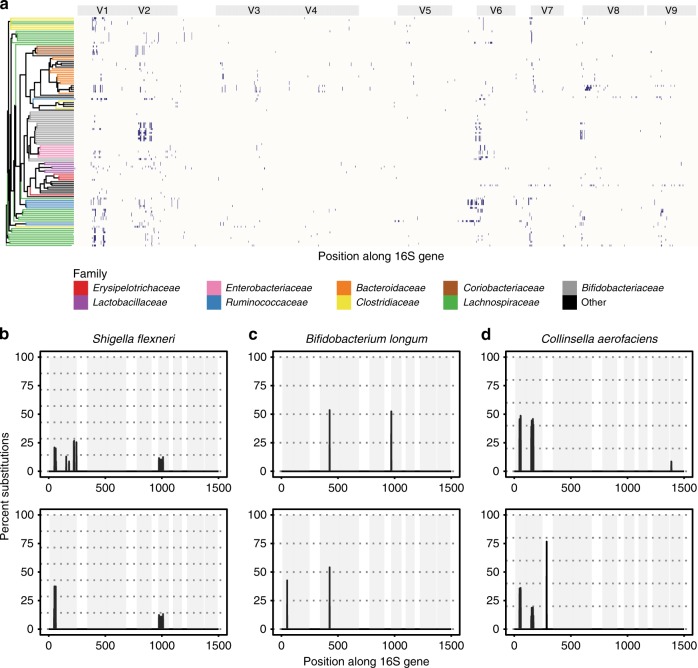
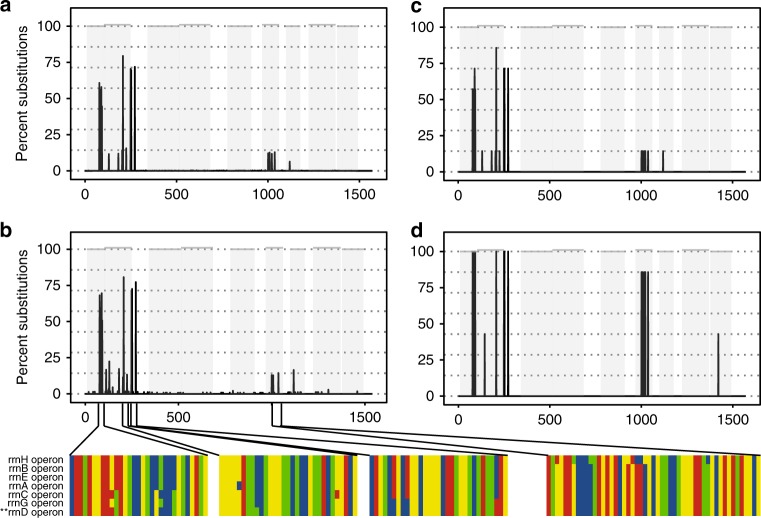
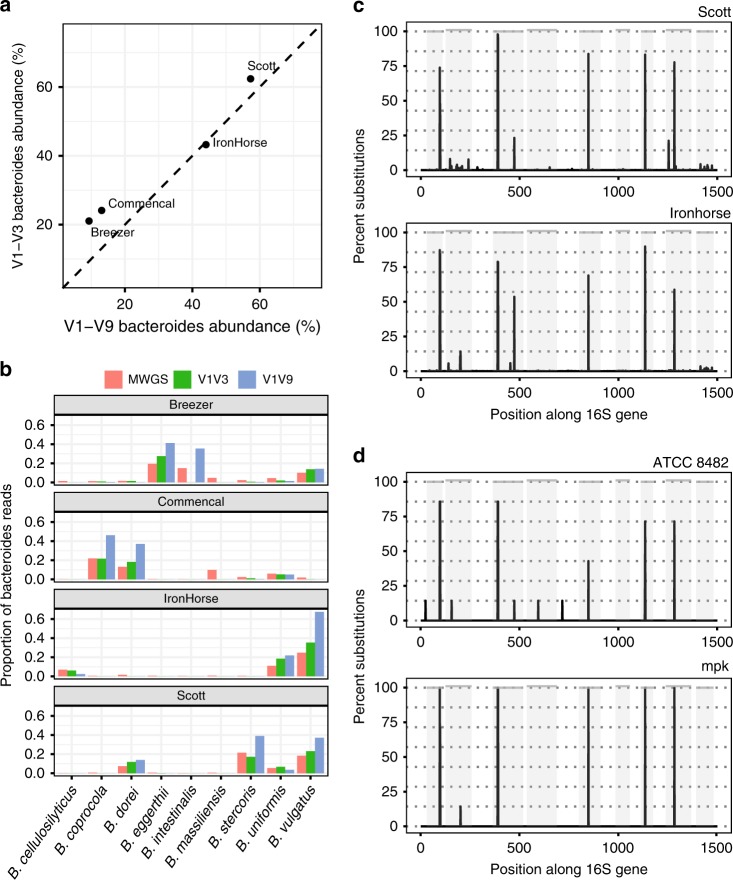
The 16S rRNA gene has been a mainstay of sequence-based bacterial analysis for decades. However, high-throughput sequencing of the full gene has only recently become a realistic prospect. Here, we use in silico and sequence-based experiments to critically re-evaluate the potential of the 16S gene to provide taxonomic resolution at species and strain level. We demonstrate that targeting of 16S variable regions with short-read sequencing platforms cannot achieve the taxonomic resolution afforded by sequencing the entire (~1500 bp) gene. We further demonstrate that full-length sequencing platforms are sufficiently accurate to resolve subtle nucleotide substitutions (but not insertions/deletions) that exist between intragenomic copies of the 16S gene. In consequence, we argue that modern analysis approaches must necessarily account for intragenomic variation between 16S gene copies. In particular, we demonstrate that appropriate treatment of full-length 16S intragenomic copy variants has the potential to provide taxonomic resolution of bacterial communities at species and strain level.
16S rRNA 基因作为一种基于序列的细菌分析方法已经有几十年的历史了。然而,全基因的高通量测序直到最近才成为一种现实的前景。在这里,我们使用计算机模拟和基于序列的实验来批判性地重新评估 16S 基因在种和菌株水平上提供分类分辨率的潜力。我们证明,使用短读测序平台靶向 16S 可变区不能实现整个(~1500bp)基因测序所提供的分类分辨率。我们进一步证明,全长测序平台足够准确,可以分辨基因组内 16S 基因拷贝之间存在的细微核苷酸取代(但不能插入/缺失)。因此,我们认为现代分析方法必须考虑到 16S 基因拷贝之间的基因组内变异。特别是,我们证明了对全长 16S 基因组内拷贝变异的适当处理有可能提供细菌群落在种和菌株水平上的分类分辨率。