Department of Biology, University of Iowa, Iowa City, IA, 52242, USA.
Iowa Neuroscience Institute, University of Iowa, Iowa City, IA, 52242, USA.
Mol Neurobiol. 2020 Feb;57(2):1146-1158. doi: 10.1007/s12035-019-01796-2. Epub 2019 Nov 7.
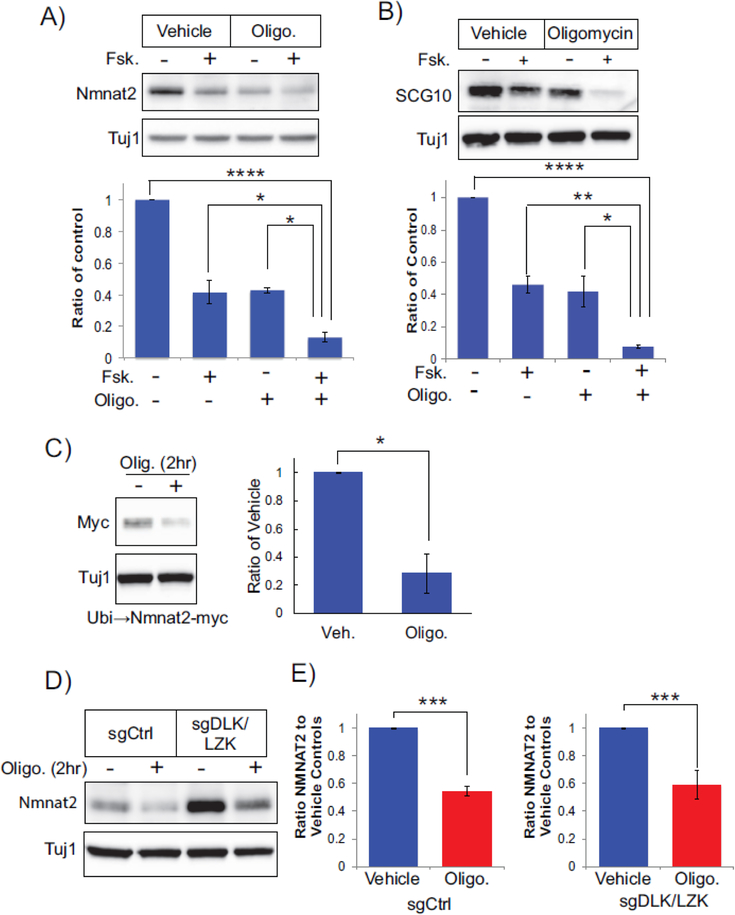
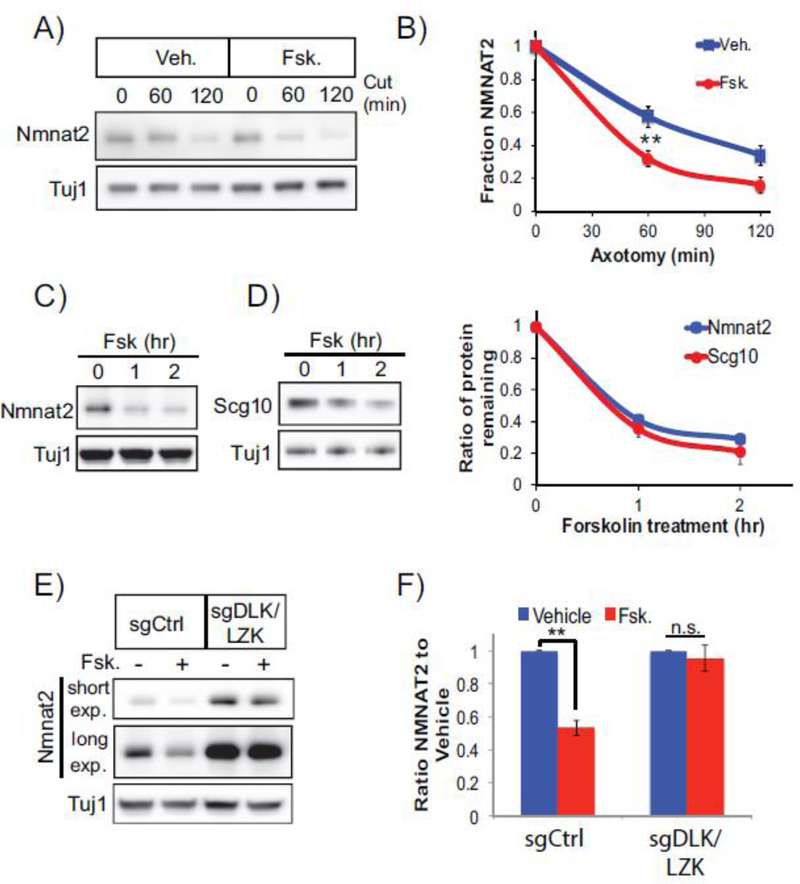
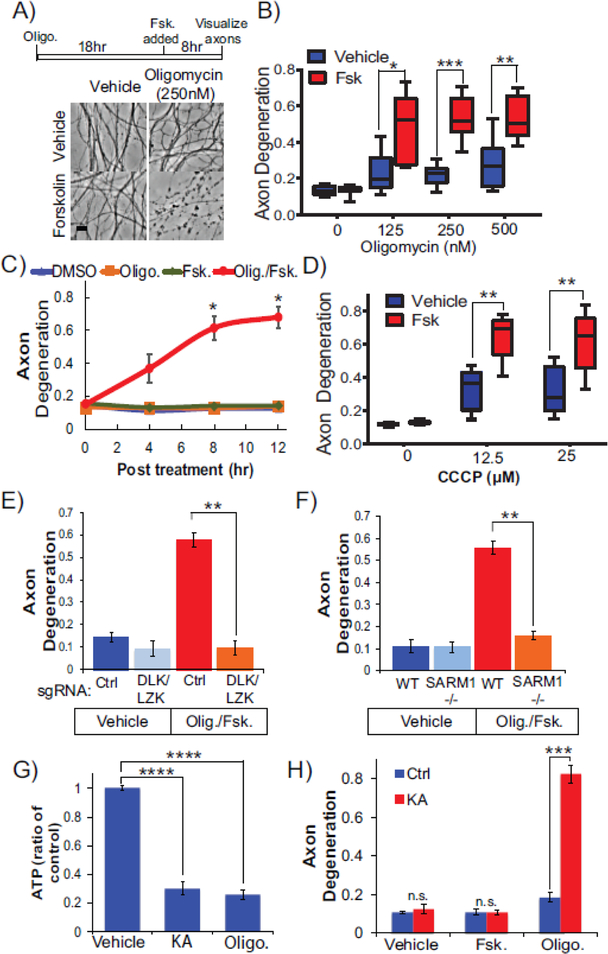
Axon degeneration is a prominent component of many neurological disorders. Identifying cellular pathways that contribute to axon vulnerability may identify new therapeutic strategies for maintenance of neural circuits. Dual leucine zipper kinase (DLK) is an axonal stress response MAP3K that is chronically activated in several neurodegenerative diseases. Activated DLK transmits an axon injury signal to the neuronal cell body to provoke transcriptional adaptations. However, the consequence of enhanced DLK signaling to axon vulnerability is unknown. We find that stimulating DLK activity predisposes axons to SARM1-dependent degeneration. Activating DLK reduces levels of the axon survival factors NMNAT2 and SCG10, accelerating their loss from severed axons. Moreover, mitochondrial dysfunction independently decreases the levels of NMNAT2 and SCG10 in axons, and in conjunction with DLK activation, leads to a dramatic loss of axonal NMNAT2 and SCG10 and evokes spontaneous axon degeneration. Hence, enhanced DLK activity reduces axon survival factor abundance and renders axons more susceptible to trauma and metabolic insult.
轴突变性是许多神经紊乱的一个突出组成部分。确定导致轴突脆弱的细胞途径可能为维持神经回路确定新的治疗策略。双亮氨酸拉链激酶 (DLK) 是一种轴突应激反应 MAP3K,在几种神经退行性疾病中持续激活。激活的 DLK 将轴突损伤信号传递到神经元细胞体,引发转录适应性。然而,增强的 DLK 信号对轴突脆弱性的后果尚不清楚。我们发现,刺激 DLK 活性使轴突容易发生 SARM1 依赖性变性。激活 DLK 降低了轴突存活因子 NMNAT2 和 SCG10 的水平,加速了它们从切断的轴突中丢失。此外,线粒体功能障碍独立地降低了轴突中 NMNAT2 和 SCG10 的水平,并且与 DLK 激活一起,导致轴突 NMNAT2 和 SCG10 的大量丢失,并引发自发轴突变性。因此,增强的 DLK 活性降低了轴突存活因子的丰度,使轴突更容易受到创伤和代谢损伤的影响。