Research Unit in Bioinformatics (RUBi), Department of Biochemistry and Microbiology, Rhodes University, Grahamstown 6140, South Africa.
Molecules. 2019 Nov 7;24(22):4036. doi: 10.3390/molecules24224036.
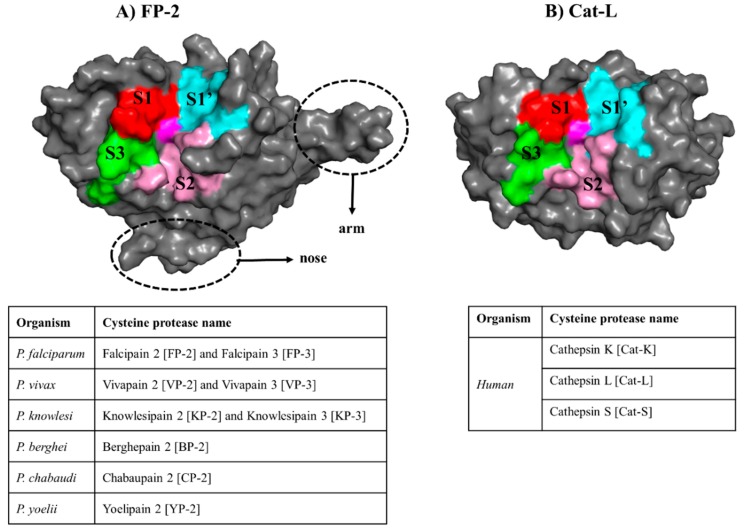
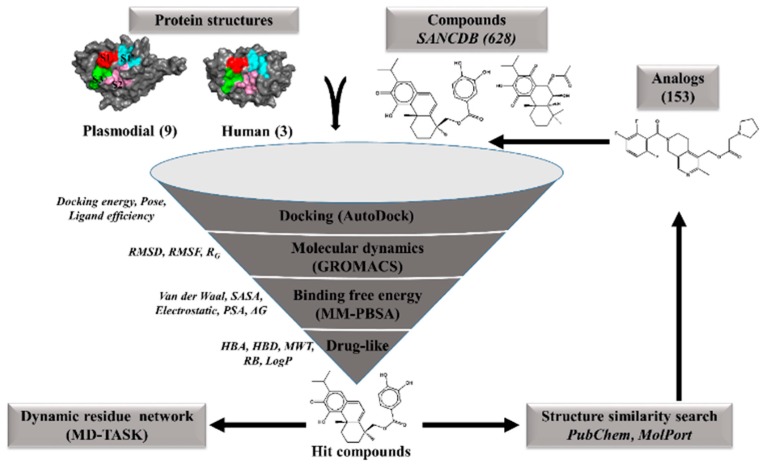
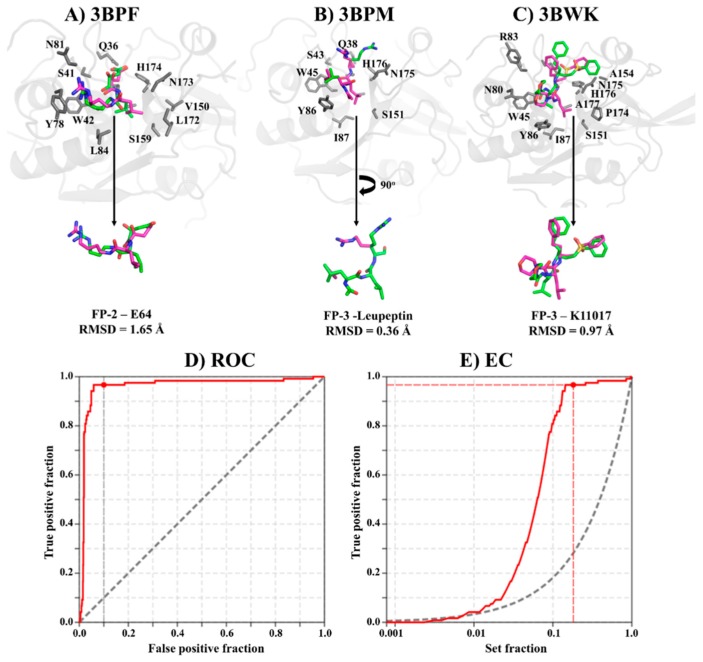
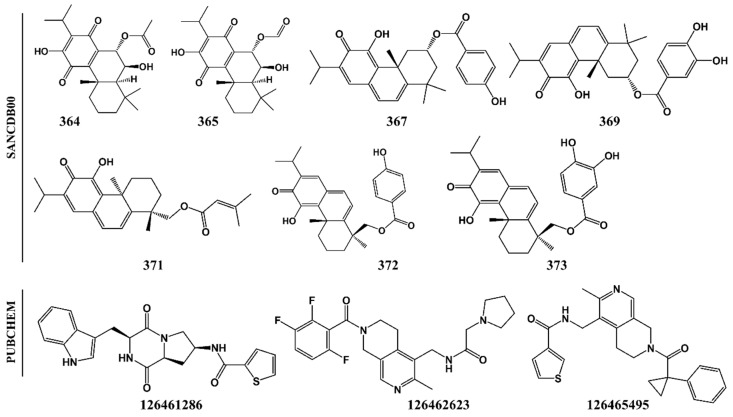
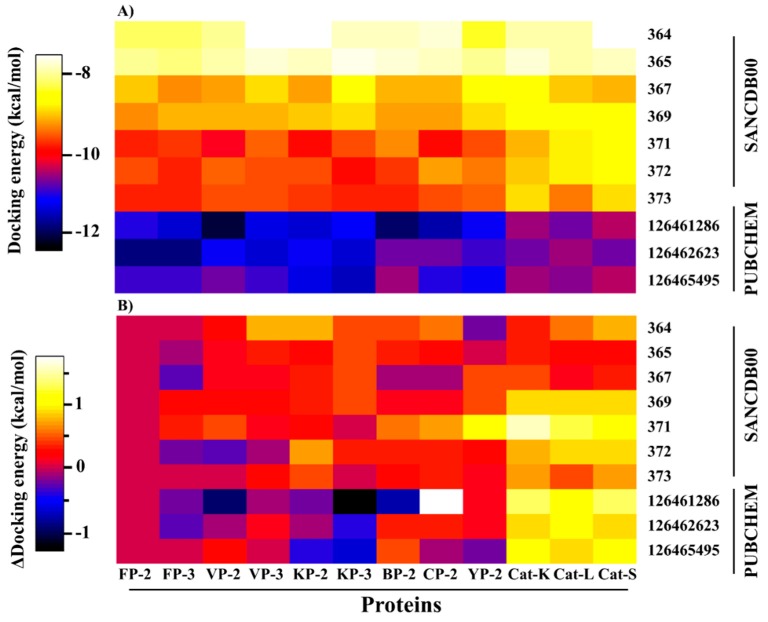
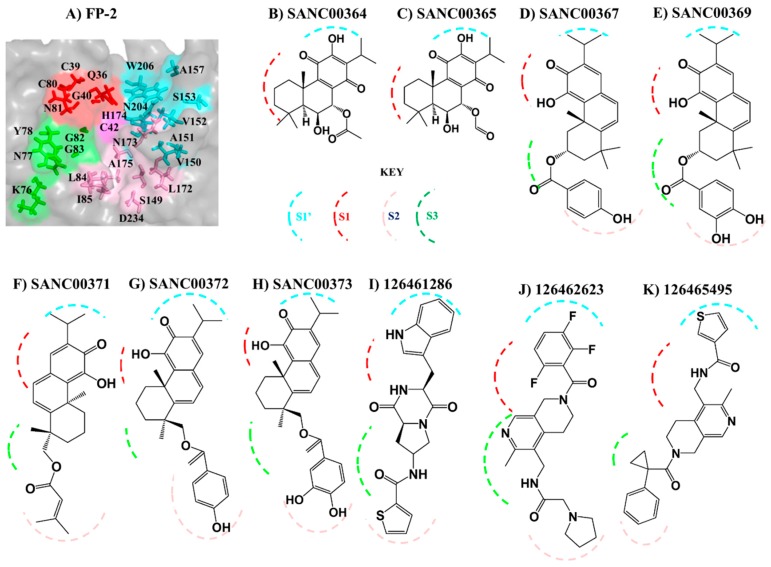
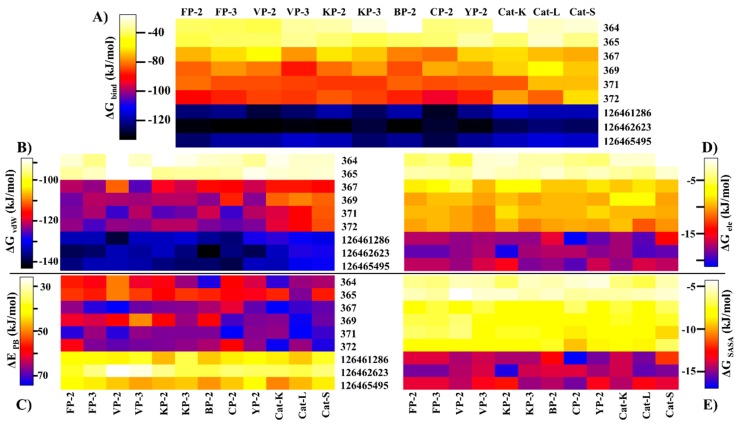
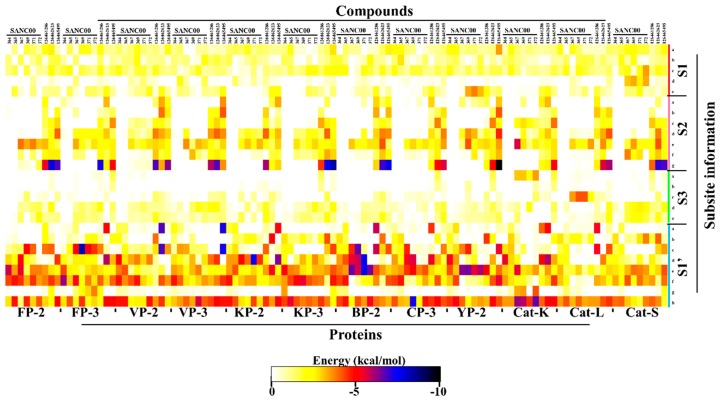
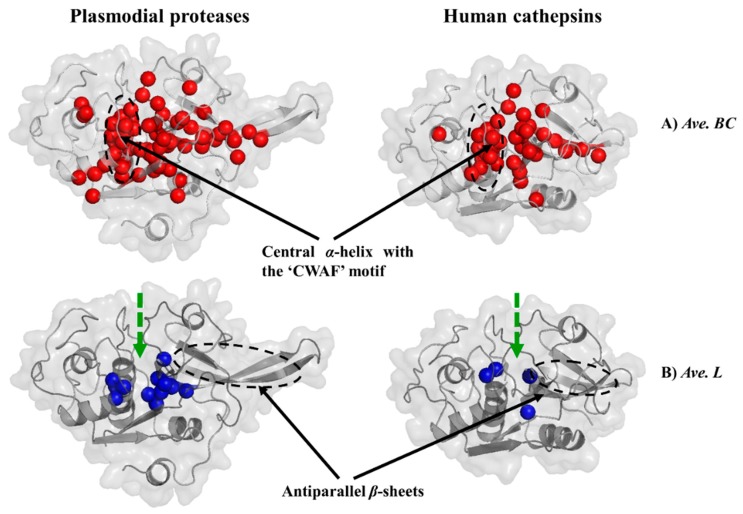
The hemoglobin degradation process in parasites is vital for nutrient acquisition required for their growth and proliferation. In , falcipains (FP-2 and FP-3) are the major hemoglobinases, and remain attractive antimalarial drug targets. Other species also possess highly homologous proteins to FP-2 and FP-3. Although several inhibitors have been designed against these proteins, none has been commercialized due to associated toxicity on human cathepsins (Cat-K, Cat-L and Cat-S). Despite the two enzyme groups sharing a common structural fold and catalytic mechanism, distinct active site variations have been identified, and can be exploited for drug development. Here, we utilize in silico approaches to screen 628 compounds from the South African natural sources to identify potential hits that can selectively inhibit the plasmodial proteases. Using docking studies, seven abietane diterpenoids, binding strongly to the plasmodial proteases, and three additional analogs from PubChem were identified. Important residues involved in ligand stabilization were identified for all potential hits through binding pose analysis and their energetic contribution determined by binding free energy calculations. The identified compounds present important scaffolds that could be further developed as plasmodial protease inhibitors. Previous laboratory assays showed the effect of the seven diterpenoids as antimalarials. Here, for the first time, we demonstrate that their possible mechanism of action could be by interacting with falcipains and their plasmodial homologs. Dynamic residue network (DRN) analysis on the plasmodial proteases identified functionally important residues, including a region with high , which had previously been proposed as a potential allosteric site in FP-2.
寄生虫中的血红蛋白降解过程对于其生长和增殖所需的营养物质获取至关重要。在疟原虫中,裂殖体蛋白(FP-2 和 FP-3)是主要的血红蛋白酶,仍然是有吸引力的抗疟药物靶点。其他疟原虫物种也具有与 FP-2 和 FP-3 高度同源的蛋白质。尽管已经针对这些蛋白质设计了几种抑制剂,但由于对人类组织蛋白酶(Cat-K、Cat-L 和 Cat-S)的相关毒性,没有一种抑制剂被商业化。尽管这两个酶组具有共同的结构折叠和催化机制,但已经确定了不同的活性位点变化,可以用于药物开发。在这里,我们利用计算方法从南非天然来源筛选了 628 种化合物,以鉴定能够选择性抑制疟原虫蛋白酶的潜在药物。通过对接研究,鉴定了七种松香烷二萜类化合物,它们与疟原虫蛋白酶结合牢固,并且从 PubChem 中鉴定了另外三种类似物。通过结合构象分析确定了所有潜在药物的配体稳定的重要残基,并通过结合自由能计算确定了它们的能量贡献。鉴定的化合物提供了重要的支架,可以进一步开发为疟原虫蛋白酶抑制剂。以前的实验室研究表明,这七种二萜类化合物具有抗疟作用。在这里,我们首次证明,它们的作用机制可能是通过与裂殖体蛋白及其疟原虫同源物相互作用。对疟原虫蛋白酶的动态残基网络(DRN)分析确定了功能重要的残基,包括一个高的区域,先前已被提议为 FP-2 中的一个潜在变构位点。