National Engineering Laboratory for Animal Breeding, Laboratory of Animal Genetics, Breeding and Reproduction, Ministry of Agriculture, College of Animal Science and Technology, China Agricultural University, Beijing, 100193, China.
Shandong Provincial Key Laboratory of Animal Biotechnology and Disease Control and Prevention, College of Animal Science and Technology, Shandong Agricultural University, Taian, 271001, China.
BMC Bioinformatics. 2019 Nov 8;20(1):556. doi: 10.1186/s12859-019-3164-z.
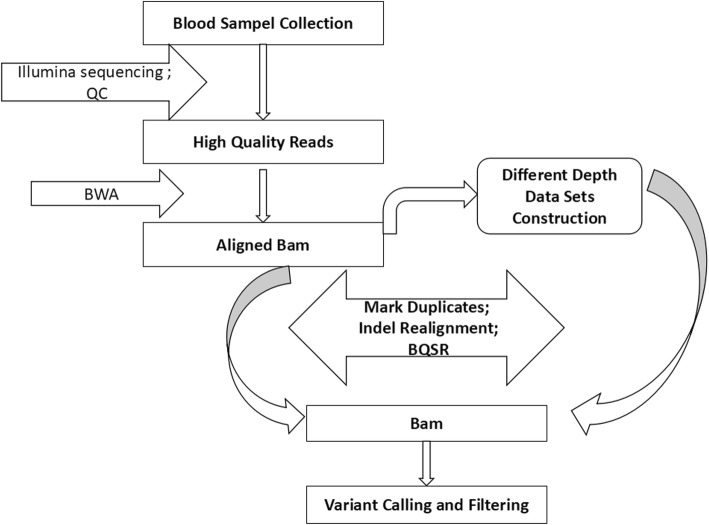
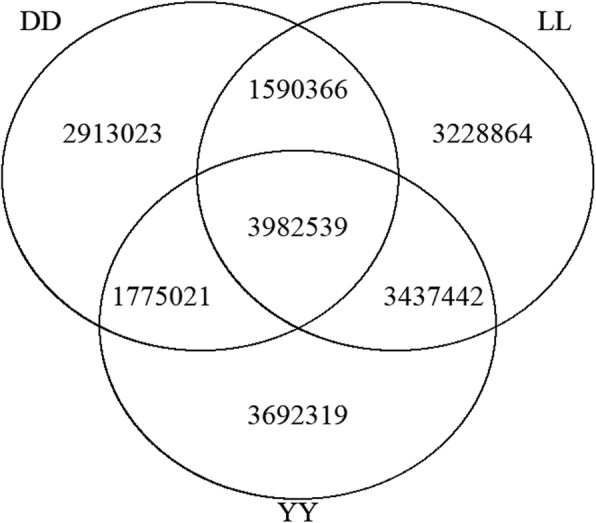
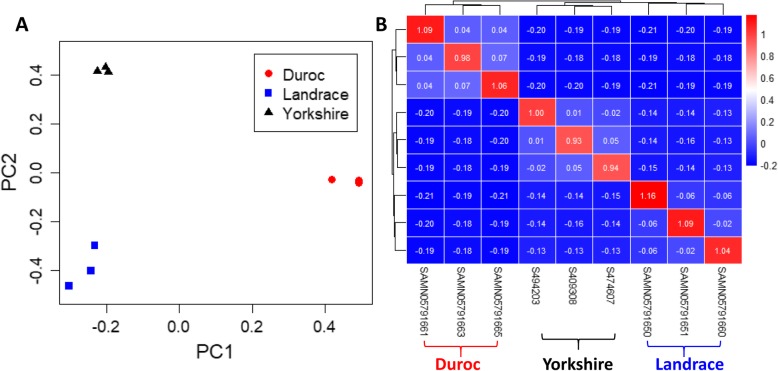
As whole-genome sequencing is becoming a routine technique, it is important to identify a cost-effective depth of sequencing for such studies. However, the relationship between sequencing depth and biological results from the aspects of whole-genome coverage, variant discovery power and the quality of variants is unclear, especially in pigs. We sequenced the genomes of three Yorkshire boars at an approximately 20X depth on the Illumina HiSeq X Ten platform and downloaded whole-genome sequencing data for three Duroc and three Landrace pigs with an approximately 20X depth for each individual. Then, we downsampled the deep genome data by extracting twelve different proportions of 0.05, 0.1, 0.15, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8 and 0.9 paired reads from the original bam files to mimic the sequence data of the same individuals at sequencing depths of 1.09X, 2.18X, 3.26X, 4.35X, 6.53X, 8.70X, 10.88X, 13.05X, 15.22X, 17.40X, 19.57X and 21.75X to evaluate the influence of genome coverage, the variant discovery rate and genotyping accuracy as a function of sequencing depth. In addition, SNP chip data for Yorkshire pigs were used as a validation for the comparison of single-sample calling and multisample calling algorithms.
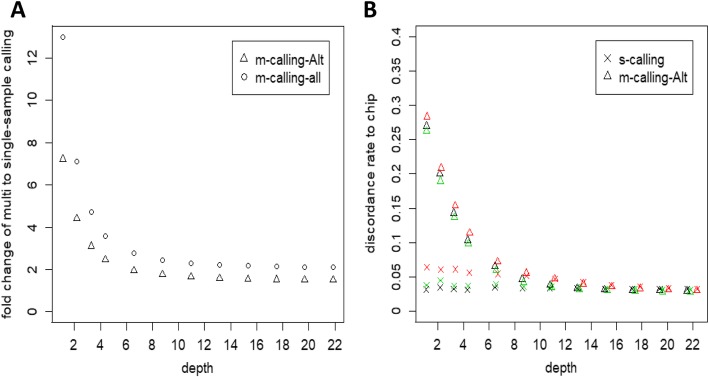
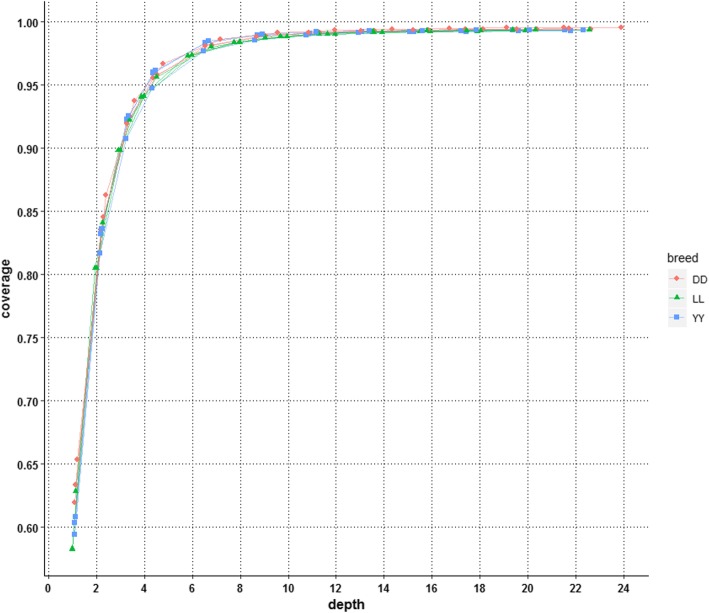
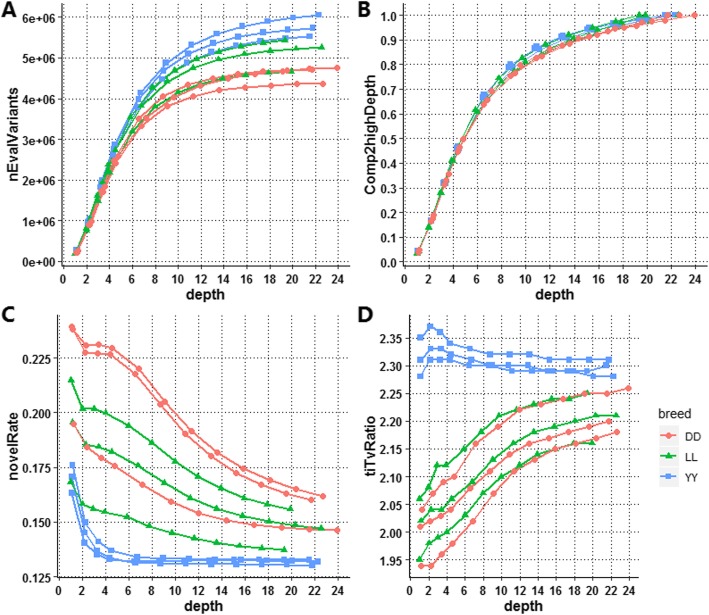
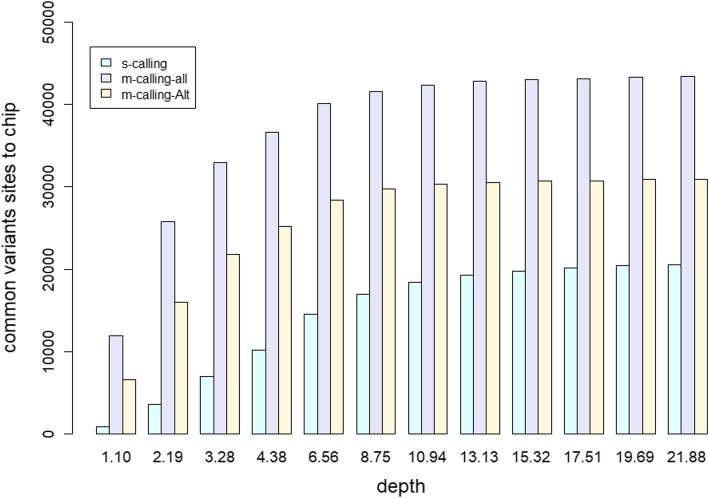
Our results indicated that 10X is an ideal practical depth for achieving plateau coverage and discovering accurate variants, which achieved greater than 99% genome coverage. The number of false-positive variants was increased dramatically at a depth of less than 4X, which covered 95% of the whole genome. In addition, the comparison of multi- and single-sample calling showed that multisample calling was more sensitive than single-sample calling, especially at lower depths. The number of variants discovered under multisample calling was 13-fold and 2-fold higher than that under single-sample calling at 1X and 22X, respectively. A large difference was observed when the depth was less than 4.38X. However, more false-positive variants were detected under multisample calling.
Our research will inform important study design decisions regarding whole-genome sequencing depth. Our results will be helpful for choosing the appropriate depth to achieve the same power for studies performed under limited budgets.
随着全基因组测序成为一种常规技术,确定这种研究的经济有效的测序深度非常重要。然而,从全基因组覆盖度、变异发现能力和变异质量等方面来看,测序深度与生物学结果之间的关系尚不清楚,尤其是在猪中。我们在 Illumina HiSeq X Ten 平台上对 3 头约克夏猪进行了大约 20X 的基因组测序,并为 3 头杜洛克猪和 3 头长白猪下载了每个个体大约 20X 的全基因组测序数据。然后,我们通过从原始 bam 文件中提取 0.05、0.1、0.15、0.2、0.3、0.4、0.5、0.6、0.7、0.8 和 0.9 对配对读取的 12 种不同比例,从原始 bam 文件中提取 12 种不同比例的 0.05、0.1、0.15、0.2、0.3、0.4、0.5、0.6、0.7、0.8 和 0.9 对配对读取,模拟相同个体在测序深度为 1.09X、2.18X、3.26X、4.35X、6.53X、8.70X、10.88X、13.05X、15.22X、17.40X、19.57X 和 21.75X 时的序列数据,以评估基因组覆盖度、变异发现率和基因分型准确性随测序深度的变化。此外,还使用约克夏猪的 SNP 芯片数据对单样本调用和多样本调用算法的比较进行了验证。
我们的结果表明,10X 是实现平台覆盖度和发现准确变异的理想实用深度,达到了大于 99%的基因组覆盖度。在深度小于 4X 时,假阳性变异的数量显著增加,覆盖了整个基因组的 95%。此外,多样本调用和单样本调用的比较表明,多样本调用比单样本调用更敏感,尤其是在较低的深度下。在 1X 和 22X 时,多样本调用发现的变异数量分别比单样本调用高 13 倍和 2 倍。在深度小于 4.38X 时,差异较大。然而,多样本调用检测到的假阳性变异数量更多。
我们的研究将为全基因组测序深度的重要研究设计决策提供信息。我们的结果将有助于选择在有限预算下实现相同研究能力的适当深度。