Department of Sciences for Health Promotion and Mother and Child Care "G. D'Alessandro", University of Palermo, Palermo, Italy.
Laboratory of Neurogenetics and Neuroscience, Institute G. Gaslini, Genoa, Italy.
Ital J Pediatr. 2019 Nov 8;45(1):138. doi: 10.1186/s13052-019-0718-7.
17q11.2 microdeletions, which include the neurofibromatosis type 1 (NF1) gene region, are responsible for the NF1 microdeletion syndrome, observed in 4.2% of all NF1 patients. Large deletions of the NF1 gene and its flanking regions are associated with a more severe NF1 phenotype than the NF1 general population.
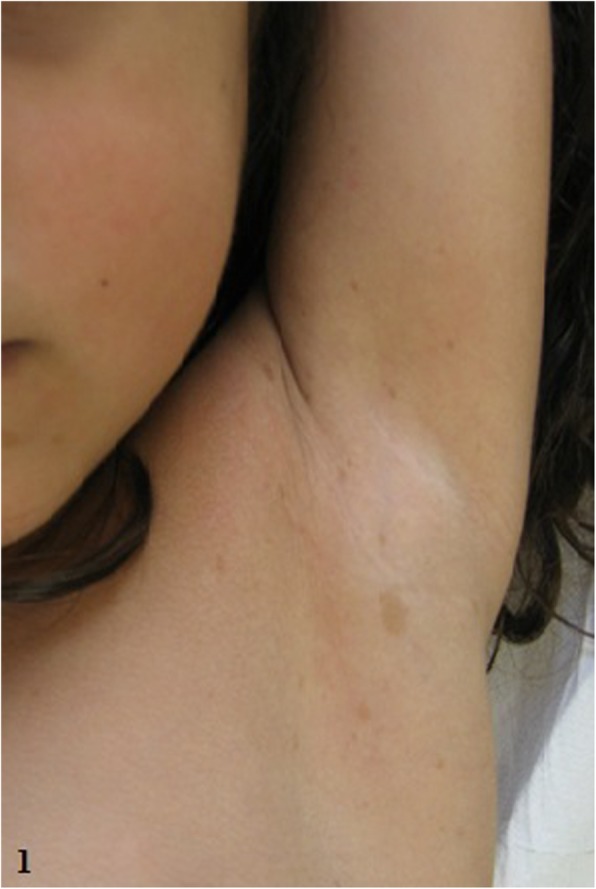
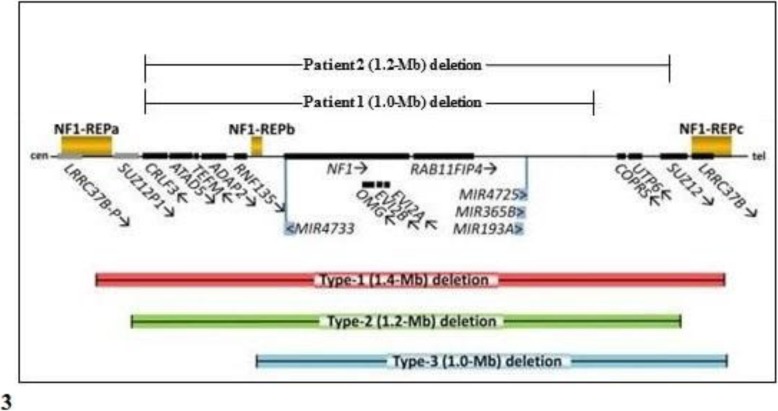
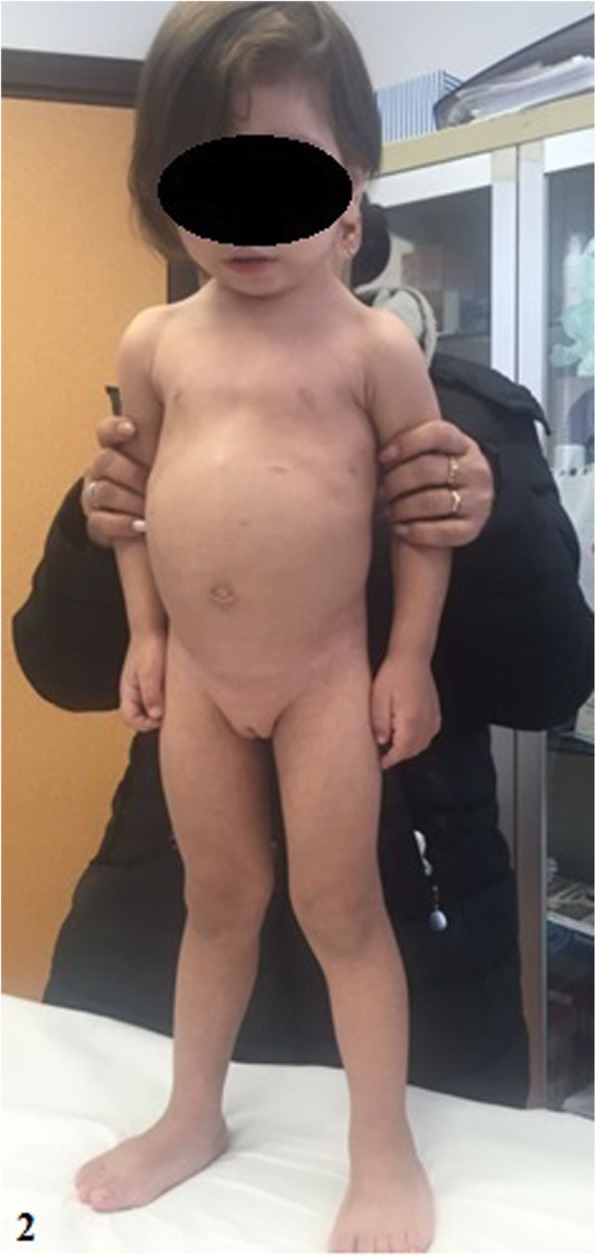
We hereby describe the clinical and molecular features of two girls (aged 2 and 4 years, respectively), with non-mosaic atypical deletions. Patient 1 showed fifteen café-au-lait spots and axillary freckling, as well as a Lisch nodule in the left eye, strabismus, high-arched palate, malocclusion, severe kyphoscoliosis, bilateral calcaneovalgus foot, mild generalized hypotonia, hyperactivity and deficits of speech-related abilities. NF1 genomic rearrangements through multiplex ligation-dependent probe amplification (MLPA) detected an heterozygous deletion of the whole NF1 gene. Array comparative genomic hybridization (a-CGH) analysis defined a 17q11.2 deletion of about 1 Mb (breakpoints at positions 29,124,299 and 30,151,654), which involved different genes (partially CRLF3, ATAD5, TEFM, ADAP2, RNF135, OMG, EVI2B, EVI2A, RAB11FIP4), including NF1. Patient 2 showed growth and developmental delay, supravalvular pulmonary stenosis, twenty-five café-au-lait spots, axillary freckling, craniofacial dysmorphic features, short neck with pterygium, limb abnormalities and foci of neural dysplasia on brain magnetic resonance imaging (MRI). MLPA detected an heterozygous deletion of NF1, which was detailed by a-CGH indicating the positions 29,124,299 and 30,326,958 as its breakpoints, and which included aside from the genes deleted in Patient 1 also COPRS, UTP6 and partially SUZ12. Fluorescent in situ hybridization (FISH) analysis of the parents documented a de novo origin of the deletions in both cases.
The present report will likely provide further insights and a better characterization of NF1 microdeletion syndrome.
17q11.2 微缺失,包括神经纤维瘤病 1 型(NF1)基因区域,是 NF1 微缺失综合征的原因,在所有 NF1 患者中占 4.2%。NF1 基因及其侧翼区域的大片段缺失与 NF1 人群中更为严重的 NF1 表型相关。
我们在此描述了两名非镶嵌性非典型缺失的女孩(分别为 2 岁和 4 岁)的临床和分子特征。患者 1 表现为 15 个咖啡牛奶斑和腋窝雀斑,左眼有 Lisch 结节,斜视,高拱形腭,牙列不齐,严重的脊柱后凸侧凸,双侧跟骨内翻足,轻度全身张力减退,多动和言语相关能力缺陷。通过多重连接依赖性探针扩增(MLPA)检测到 NF1 基因组重排存在 NF1 基因的杂合性缺失。阵列比较基因组杂交(a-CGH)分析定义了一个约 1Mb 的 17q11.2 缺失(断点位于 29,124,299 和 30,151,654 位置),该缺失涉及不同的基因(部分包括 CRLF3、ATAD5、TEFM、ADAP2、RNF135、OMG、EVI2B、EVI2A、RAB11FIP4),包括 NF1。患者 2 表现为生长发育迟缓,瓣上型肺动脉狭窄,25 个咖啡牛奶斑,腋窝雀斑,颅面畸形特征,短颈伴翼状胬肉,肢体异常和脑磁共振成像(MRI)上的神经发育不良灶。MLPA 检测到 NF1 的杂合性缺失,a-CGH 详细说明了其断点位于 29,124,299 和 30,326,958 位置,除了患者 1 缺失的基因外,还包括 COPRS、UTP6 和部分 SUZ12。对父母的荧光原位杂交(FISH)分析记录了两个病例中缺失的新生来源。
本报告可能会提供更多关于 NF1 微缺失综合征的见解和更好的特征描述。