Institute for Molecular Bioscience, University of Queensland, St Lucia, Brisbane, 4072, QLD, Australia.
Estonian Genome Center, Institute of Genomics, University of Tartu, Riia 23b, 51010, Tartu, Estonia.
Nat Commun. 2019 Nov 8;10(1):5086. doi: 10.1038/s41467-019-12653-0.
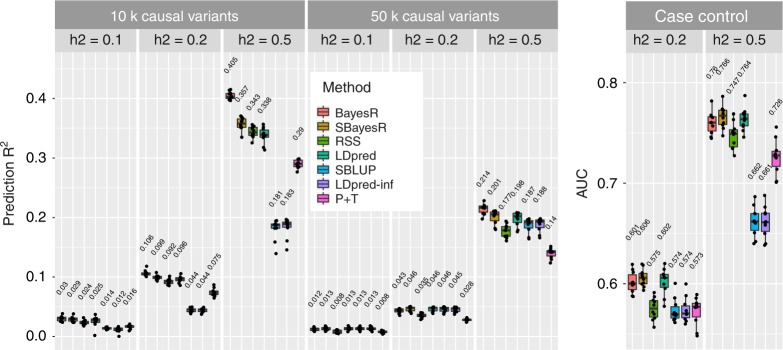
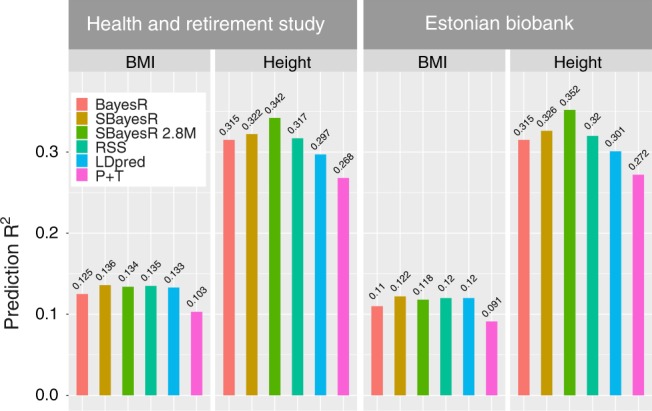
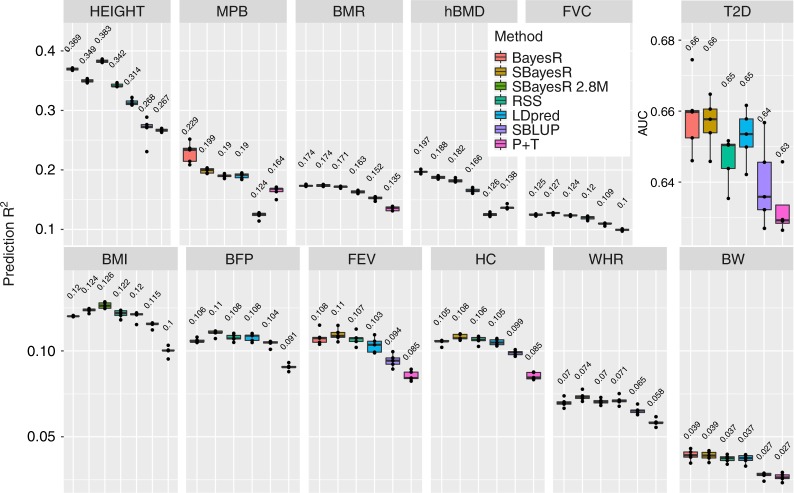
Accurate prediction of an individual's phenotype from their DNA sequence is one of the great promises of genomics and precision medicine. We extend a powerful individual-level data Bayesian multiple regression model (BayesR) to one that utilises summary statistics from genome-wide association studies (GWAS), SBayesR. In simulation and cross-validation using 12 real traits and 1.1 million variants on 350,000 individuals from the UK Biobank, SBayesR improves prediction accuracy relative to commonly used state-of-the-art summary statistics methods at a fraction of the computational resources. Furthermore, using summary statistics for variants from the largest GWAS meta-analysis (n ≈ 700, 000) on height and BMI, we show that on average across traits and two independent data sets that SBayesR improves prediction R by 5.2% relative to LDpred and by 26.5% relative to clumping and p value thresholding.
从个体的 DNA 序列准确预测其表型是基因组学和精准医学的重大承诺之一。我们将强大的个体水平数据贝叶斯多元回归模型(BayesR)扩展为一种利用全基因组关联研究(GWAS)汇总统计数据的模型(SBayesR)。在使用来自英国生物库的 35 万名个体的 12 个真实特征和 110 万个变体进行的模拟和交叉验证中,SBayesR 提高了预测准确性,而计算资源仅为常用的最先进汇总统计数据方法的一小部分。此外,使用来自最大 GWAS 荟萃分析(n≈700000)的身高和 BMI 变体的汇总统计数据,我们表明,在跨特征和两个独立数据集的情况下,SBayesR 平均将预测 R 提高了 5.2%,与 LDpred 相比提高了 26.5%,与聚类和 p 值阈值相比提高了 26.5%。