Behjati Ardakani Fatemeh, Schmidt Florian, Schulz Marcel H
High throughput Genomics and Systems Biology, Cluster of Excellence on Multimodel Computing and Interaction, Saarland University, Saarbruecken,, Saarland, 66123, Germany.
Computational Biology and Applied Algorithmics, Max Planck Institute for Informatics, Saarbruecken, Saarland, 66123, Germany.
F1000Res. 2018 Oct 4;7:1603. doi: 10.12688/f1000research.16200.2. eCollection 2018.
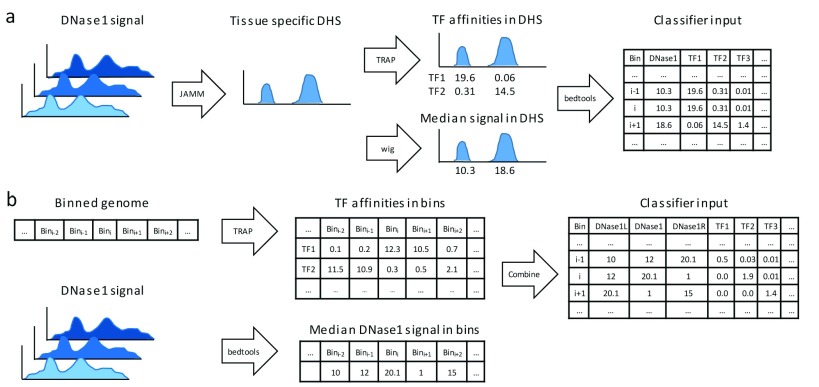
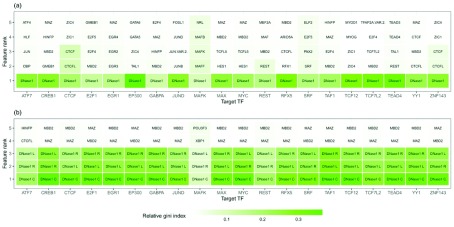
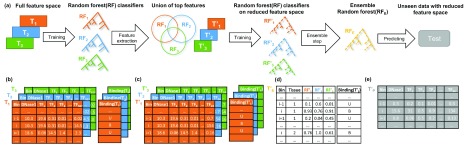
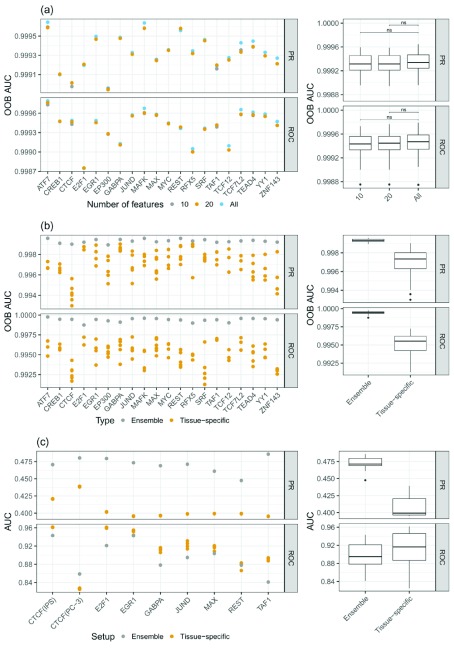
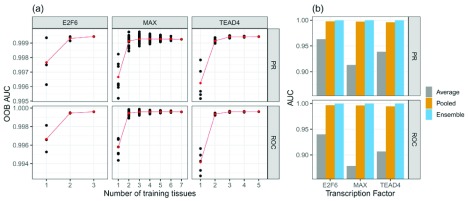
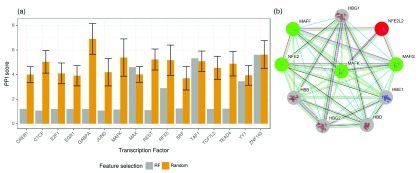
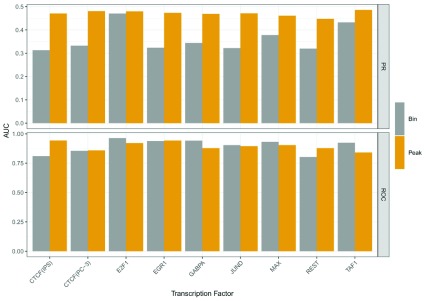
: Understanding the location and cell-type specific binding of Transcription Factors (TFs) is important in the study of gene regulation. Computational prediction of TF binding sites is challenging, because TFs often bind only to short DNA motifs and cell-type specific co-factors may work together with the same TF to determine binding. Here, we consider the problem of learning a general model for the prediction of TF binding using DNase1-seq data and TF motif description in form of position specific energy matrices (PSEMs). We use TF ChIP-seq data as a gold-standard for model training and evaluation. Our contribution is a novel ensemble learning approach using random forest classifiers. In the context of the we consider different learning setups. Our results indicate that the ensemble learning approach is able to better generalize across tissues and cell-types compared to individual tissue-specific classifiers or a classifier built based upon data aggregated across tissues. Furthermore, we show that incorporating DNase1-seq peaks is essential to reduce the false positive rate of TF binding predictions compared to considering the raw DNase1 signal. Analysis of important features reveals that the models preferentially select motifs of other TFs that are close interaction partners in existing protein protein-interaction networks. Code generated in the scope of this project is available on GitHub: https://github.com/SchulzLab/TFAnalysis (DOI: 10.5281/zenodo.1409697).
了解转录因子(TFs)的定位和细胞类型特异性结合在基因调控研究中很重要。TF结合位点的计算预测具有挑战性,因为TF通常仅与短DNA基序结合,并且细胞类型特异性辅助因子可能与同一TF协同作用以确定结合。在这里,我们考虑使用DNase1-seq数据和以位置特异性能量矩阵(PSEM)形式的TF基序描述来学习用于预测TF结合的通用模型的问题。我们使用TF ChIP-seq数据作为模型训练和评估的金标准。我们的贡献是一种使用随机森林分类器的新型集成学习方法。在本文的背景下,我们考虑了不同的学习设置。我们的结果表明,与单个组织特异性分类器或基于跨组织聚合数据构建的分类器相比,集成学习方法能够更好地在不同组织和细胞类型中进行泛化。此外,我们表明,与考虑原始DNase1信号相比,纳入DNase1-seq峰对于降低TF结合预测的假阳性率至关重要。对重要特征的分析表明,模型优先选择在现有蛋白质-蛋白质相互作用网络中作为紧密相互作用伙伴的其他TF的基序。本项目范围内生成的代码可在GitHub上获取:https://github.com/SchulzLab/TFAnalysis(DOI:10.5281/zenodo.1409697)。