Victor Chang Cardiac Research Institute, Darlinghurst, NSW, 2010, Australia.
St. Vincent's Clinical School, University of New South Wales, Darlinghurst, NSW, 2010, Australia.
BMC Genomics. 2018 Jan 19;19(Suppl 1):929. doi: 10.1186/s12864-017-4340-z.
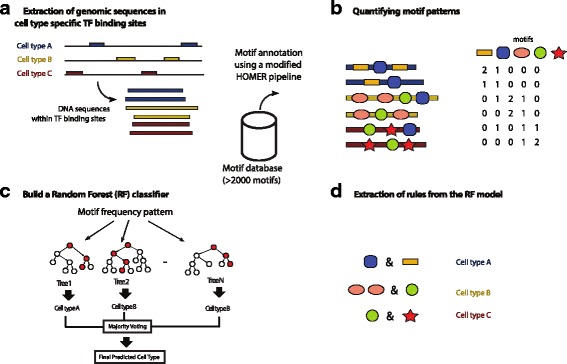
It has been observed that many transcription factors (TFs) can bind to different genomic loci depending on the cell type in which a TF is expressed in, even though the individual TF usually binds to the same core motif in different cell types. How a TF can bind to the genome in such a highly cell-type specific manner, is a critical research question. One hypothesis is that a TF requires co-binding of different TFs in different cell types. If this is the case, it may be possible to observe different combinations of TF motifs - a motif grammar - located at the TF binding sites in different cell types. In this study, we develop a bioinformatics method to systematically identify DNA motifs in TF binding sites across multiple cell types based on published ChIP-seq data, and address two questions: (1) can we build a machine learning classifier to predict cell-type specificity based on motif combinations alone, and (2) can we extract meaningful cell-type specific motif grammars from this classifier model.
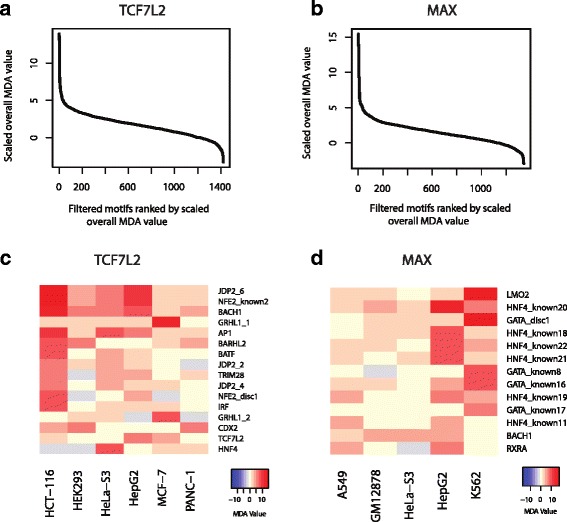
We present a Random Forest (RF) based approach to build a multi-class classifier to predict the cell-type specificity of a TF binding site given its motif content. We applied this RF classifier to two published ChIP-seq datasets of TF (TCF7L2 and MAX) across multiple cell types. Using cross-validation, we show that motif combinations alone are indeed predictive of cell types. Furthermore, we present a rule mining approach to extract the most discriminatory rules in the RF classifier, thus allowing us to discover the underlying cell-type specific motif grammar.
Our bioinformatics analysis supports the hypothesis that combinatorial TF motif patterns are cell-type specific.
已经观察到,许多转录因子(TFs)可以根据表达 TF 的细胞类型而结合到不同的基因组位置,即使单个 TF 通常在不同的细胞类型中结合到相同的核心基序。TF 如何以如此高度的细胞类型特异性方式结合到基因组,是一个关键的研究问题。一种假设是,TF 需要在不同的细胞类型中共同结合不同的 TF。如果是这样,那么可能可以观察到不同的 TF 基序组合 - 一种基序语法 - 位于不同细胞类型中的 TF 结合位点。在这项研究中,我们开发了一种基于已发表的 ChIP-seq 数据的生物信息学方法,用于系统地识别多个细胞类型中 TF 结合位点的 DNA 基序,并解决两个问题:(1)我们能否仅基于基序组合构建一种机器学习分类器来预测细胞类型特异性,以及(2)我们能否从该分类器模型中提取有意义的细胞类型特异性基序语法。
我们提出了一种基于随机森林(RF)的方法来构建多类分类器,以根据其基序内容预测 TF 结合位点的细胞类型特异性。我们将这种 RF 分类器应用于两个已发表的跨多个细胞类型的 TF(TCF7L2 和 MAX)的 ChIP-seq 数据集。通过交叉验证,我们表明基序组合本身确实可以预测细胞类型。此外,我们提出了一种规则挖掘方法来提取 RF 分类器中最具区分性的规则,从而使我们能够发现潜在的细胞类型特异性基序语法。
我们的生物信息学分析支持这样的假设,即组合 TF 基序模式是细胞类型特异性的。